Introduction
The explosion in biological sequence information has led to the generation of large multiple sequence alignments (MSA). For example, the biggest protein family alignment currently in the Pfam database (Wellcome Trust-Sanger Institute) has over 288,000 sequences1. A new generation of alignment programs, such as Clustal Omega2, are available that allow the routine calculation of such large alignments. However, a Nature Methods review3 noted the lack of software tools for visualizing the results of large alignment calculations. Specifically, there was a call for overcoming the conceptual and technical limitations of large data sets to allow one to navigate visually both an overview and the details of an alignment, while having mechanisms to query annotated data. We point out that this conceptual limitation was solved in late 2008 by the introduction of ProfileGrids as a new paradigm for visualizing large multiple sequence alignments4. Here, we report that the remaining technical limitations have been overcome with version 2.0 of the JProfileGrid software, and that therefore, the large alignment visualization problem has now been solved. We use the influenza hemagglutinin protein family as a case study to demonstrate the new features of the software.
Previous MSA visualization paradigms3,5 do not provide both alignment overviews together with the details of each character’s frequency distribution at each homologous position in the entire alignment. A particular technical limitation of stacked sequence representations is that as the alignment size grows, a printed or digital visualization can run out of convenient space. This compounds the conceptual limitation where the user cannot grasp the overall conservation trends and the observed variation details in a large data set. The ProfileGrid paradigm was introduced as a solution to this conceptual problem by converting a multiple sequence alignment to a color-coded matrix of the residue frequency occurring at every homologous position across the entire length of a MSA. Therefore, all MSA information is represented, both at variable regions and of infrequent residues that may yield clues about biological function.
Improvements to JProfileGrid software
Comparisons of the stacked sequence and ProfileGrid visualization paradigms highlight the challenges of large MSAs (Fig. 1). We downloaded 11,981 full-length, non-redundant hemagglutinin protein sequences from the NCBI influenza virus resource6, aligned them using MUSCLE7, and visualized the MSA8 with the new JProfileGrid 2.0 software. Both paradigms depict the entire width of the alignment, i.e., 650 columns from the approximately 570 protein residues and the 80 inserted gap characters. However, due to space limitations, the stacked sequence overview (Fig. 1a) is only a sampling of 600 sequences from the whole alignment. In the stacked representation, each row is an individual protein sequence with the amino acids represented as pixels colored according to the Taylor scheme9. Below the stacked sequence overview is a similarly colored magnified view (Fig. 1b) from a much smaller alignment with the amino acid one-letter codes shown. By contrast, the ProfileGrid overview representation (Fig. 1c) divides the alignment width into multiple "tiers" of which six are 100 characters wide. Within each tier are 21 rows for the 20 amino acids and a gap character. Each cell in the matrix is a color shaded count of the character occurrence at the corresponding MSA column position from low (white) to high (dark blue) frequency. Thus, all of the sequences from the entire alignment are represented in the ProfileGrid by the frequency color shading of each cell in the matrix. As a reference, the left-most column of each ProfileGrid tier is colored according to the Taylor scheme for each amino acid row. ProfileGrids solve the visualization problem of large alignments since there is no limit to the number of sequences that can be represented. Note that a stacked sequence representation of all 11,981 hemagglutinin sequences in this example would be twenty times larger than the number of rows shown in the overview (Fig. 1a).
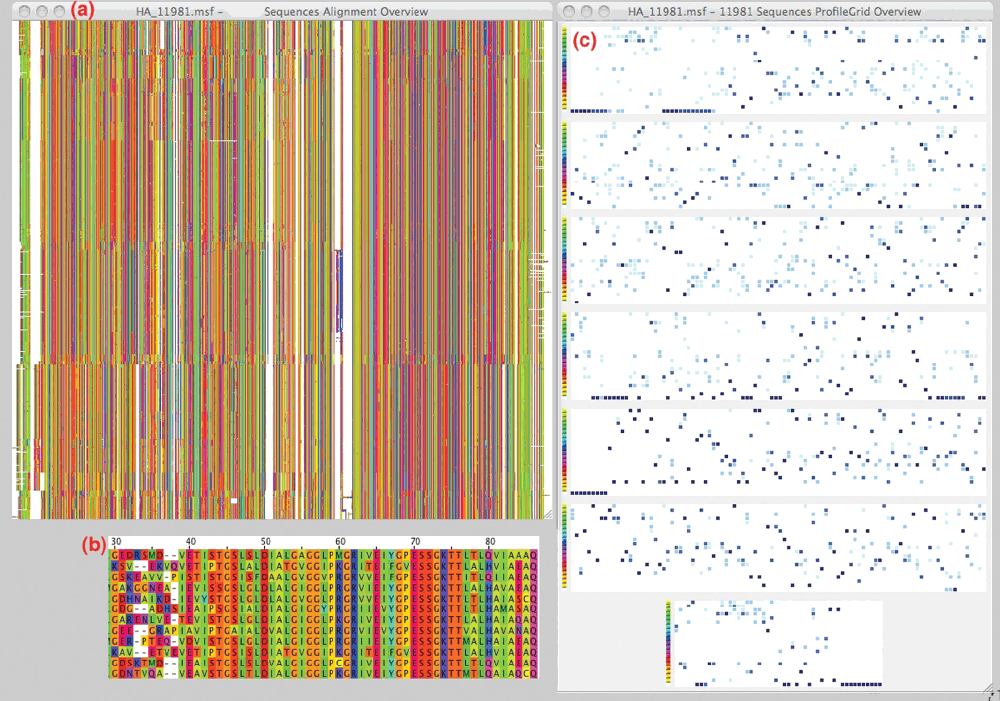
Figure 1. Stacked sequence versus ProfileGrid visualization paradigms.
The alignment of hemagglutinin sequences is compared side-by-side using the new JProfileGrid 2.0 "overview" feature for both stacked sequence (a) and ProfileGrid (c) visualization methods. An example detailed view of a stacked sequence aligment shows only 12 sequences (b) with the one-letter amino acid codes colored according to the Taylor scheme. While space limitations restrict the stacked sequence overview to only 600 sequences, the ProfileGrid overview represents the entire residue content of all 11,981 sequences. See text for details.
The first version of the Java JProfileGrid software was designed for alignments with hundreds of sequences such as the ubiquitous RecA protein involved in bacterial recombination4. Upon analyzing virus protein families such as influenza hemagglutinins with thousands of sequences, it became apparent that the larger data sets were taxing certain software technical limits. We completely overhauled the program to improve the code with respect to object oriented design, memory management, calculation efficiency, and speed. We strategically reduced memory usage and introduced parallelized code for computing amino acid frequency counts. For example, we were able to reduce the memory requirements by addressing a technical limitation of the Java programming language. Java uses 2 bytes (16-bits) of memory for every Unicode character (UTF-16) reflecting the need to support thousands of characters from hundreds of languages. However, typical MSAs consist of only ASCII characters. Thus, we were able to reduce the memory use 2-fold by introducing a byte (8-bit) map between the integers 0 to 20 and the twenty common amino acid codes and a gap character. Parallelization was possible due to the nature of the ProfileGrid format. Amino acid counts within each column are independent of each other and so were partitioned into separately running threads thereby taking advantage of multi-core processor environments for enhanced ProfileGrid calculation speed.
New features in JProfileGrid v2.0 accommodate viewing and analyzing very large MSAs. The software now allows sorting and searching the menu list of sequence names for finding a specific homolog to serve as the reference sequence for the ProfileGrid. The new "overview" modes enable the user to visualize the entire MSA within one window. The detailed ProfileGrid window with the character counts has a new second pane that facilitates simultaneous viewing of different parts of the MSA. To easily focus on rare residues, the "highlight" functionality now identifies residues that occur greater or less than a user-defined threshold of residue frequency. We introduced data sampling to accelerate similarity plot calculations rather than needlessly including every single sequence from large MSAs. Regular expression functionality has also been implemented to find sequences with particular names or amino acid patterns. Finally, JProfileGrid can import simple sequence annotations from flat file spreadsheet databases10 for metadata filtering to reduce large MSAs to subsets of interest.
Example: influenza hemagglutinin
As a demonstration of the new metadata (Fig. 2a) and regular expression (Fig. 2b) software features, the 11,981 sequence alignment (Fig. 2c) was filtered to just 60 hemagglutinin homologs (Fig. 2d) by using a metadata search for "human" hosts and a regular expression search for a "Mexico" country location. The ProfileGrid views are positioned at alignment column 333 where there is a potential glycosylation site (asn-thr-thr-cys underlined in red) that is found among this sequence subset. Glycosylation is a key post-translational modification that is vital to the proper folding and trafficking of viral proteins. In addition to the role in protein folding and processing, glycosylation in influenza is also important to virus immune evasion strategy11. The majority of antibody recognition is dedicated to the surface antigen on the globular head although stem-directed antibodies have recently been described12. Glycosylation in the stem region could prevent recognition of these antibodies and allow for virus to escape the immune response. The region visualized by ProfileGrid analysis lies in the stem region of the hemagglutinin molecule. ProfileGrids, therefore, permit observing rare natural sequence variation (Fig. 2d) within the context of an entire multiple sequence alignment (Fig. 2c). ProfileGrids have this unique advantage over other compressed alignment visualization methods such as sequence logos13.
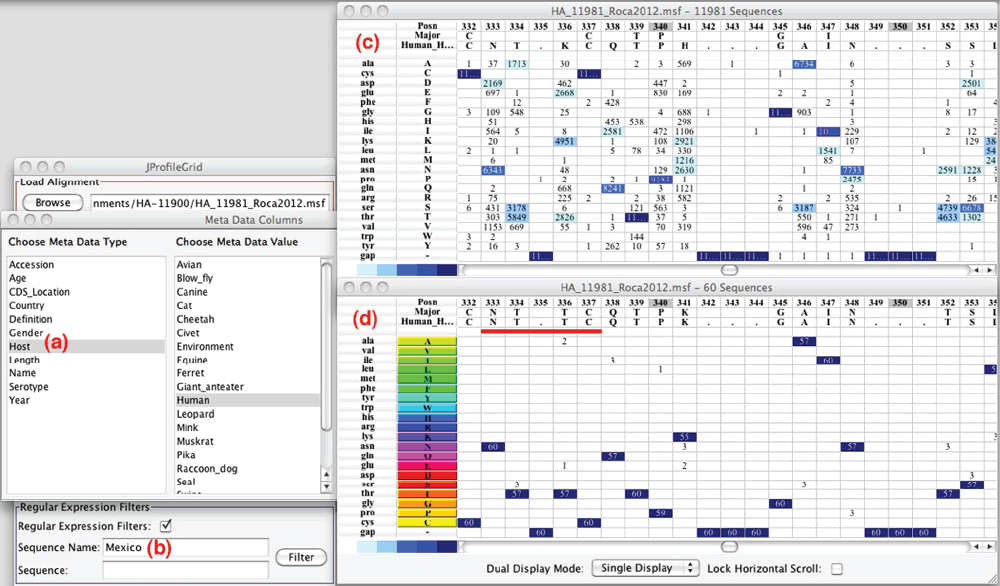
Figure 2. Screenshot of the JProfileGrid 2.0 interactive viewer and search features.
The metadata (a) and regular expression filters (b) allow the entire sequence alignment (c) to be filtered to a select subset of sequences (d). Note that in panel (d), the amino acids are colored and sorted according to the Taylor classification scheme. A glycosylation site (red underline; MSA column position 333) is located within bioinformatic element variable_5a. See text for details.
In conclusion, ProfileGrids have solved the problem of visualizing very large alignments. As sequence data sets grow, both for the end user and in central database repositories, we anticipate that ProfileGrids will simplify the dissection and analysis of MSAs. Parenthetically, we note that some bioinformatic studies about influenza proteins have lacked figures depicting the alignment upon which the analyses are based14. This omission was probably due to technical limitations that now no longer exist. Thus, we propose that ProfileGrid representations of alignment datasets be included as part of publication to assist science communication. This may initiate establishing a future standard for MIMSA: Minimum Information about a Multiple Sequence Alignment15. Likewise, database curators can use JProfileGrid and its new PNG image output to include ProfileGrid visualizations with protein family descriptions and search results. JProfileGrid v2.0 is available under a GNU General Public License and can be downloaded from http://www.ProfileGrid.org.
Author contributions
AIR and DJV conceived the study. AIR designed the software, collected hemagglutinin sequences, performed the bioinformatic analysis and biocuration, and prepared the first draft of the manuscript. ACA wrote Java code and contributed to writing the manuscript. DJV interpreted the bioinformatic observations and contributed to writing the manuscript. AIR and ACA contributed equally to the work. All authors were involved in the revision of the draft manuscript and have agreed to the final content.
Competing interests
We have developed the software but have no competing interests.
Grant information
AIR was supported by the Erasmo Foundation (grant TSC13702); and DJV was supported by a Veterans Administration Career Development Grant. This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development with resources and the use of facilities at the VA Tennessee Valley Healthcare System.
Acknowledgements
We thank David Talalayevsky and Andy Pitt for programming support; Tamara Munzner and Adam Steinberg for visualization design suggestions; and the ISMB KillerApp Award, VIZBI, and SACNAS Postdoc committees for helpful discussions.
References
- 1.
Punta M, Coggill PC, Eberhardt RY, et al:The Pfam protein families database.
Nucleic Acids Res.
(2012)40, D290–301.
- 2.
Sievers F, Wilm A, Dineen D, et al:Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega.
Mol Syst Biol,
(2011)7, 539.
- 3.
Procter JB, Thompson J, Letunic I, et al:Visualization of multiple alignments, phylogenies and gene family evolution.
Nat Methods,
(2010)7, S16–25.
- 4.
Roca AI, Almada AE, Abajian AC, et al:ProfileGrids as a new visual representation of large multiple sequence alignments: a case study of the RecA protein family.
BMC Bioinformatics,
(2008)9, 554.
- 5.
Puntervoll P, Aasland R: In Cesareni, G., Gimona, M., Sudol, M. and Yaffe, M. (eds.), Modular Protein Domains. Wiley-VCH, Weinheim, Germany, (2005), pp. 477–486.
- 6.
Bao Y, Bolotov P, Dernovoy D, et al:The influenza virus resource at the National Center for Biotechnology Information.
J Virol.
(2008)82, 596–601.
- 7.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput.
Nucleic Acids Res.
(2004)32, 1792–1797.
- 8.
Roca AI: Multiple sequence alignment of influenza hemagglutinin protein sequences.figshare, (2012).
- 9.
Taylor WR: Residual colours: a proposal for aminochromography.
Protein Eng,
(1997)10, 743–746.
- 10.
Roca AI: Meta data for multiple sequence alignment of influenza hemagglutinin protein sequences.figshare, (2012).
- 11.
Vigerust DJ, Shepherd VL: Virus glycosylation: role in virulence and immune interactions.
Trends Microbiol.
(2007)15, 211–218.
- 12.
Fleishman SJ, Whitehead TA, Ekiert DC, et al:Computational design of proteins targeting the conserved stem region of influenza hemagglutinin.
Science,
(2011)332, 816–821.
- 13.
Schneider TD, Stephens RM: Sequence logos: a new way to display consensus sequences.
Nucleic Acids Res.
(1990)18, 6097–6100.
- 14.
Amaro RE, Swift RV, Votapka L, et al:Mechanism of 150-cavity formation in influenza neuraminidase.
Nat Commun.
(2011)2, 388.
- 15.
Taylor CF, Field D, Sansone SA, et al:Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project.
Nat Biotechnol.
(2008)26, 889–896.
Comments on this article Comments (0)