Introduction
Primary synovial sarcoma arising in the lung is very rarely seen in clinical practice1,2. Before 2005, only 60 cases of synovial sarcoma in the pleuropulmonary region were reported in the English-language scientific literature3. It is normally a tumor found in adolescents and young adults between 15 and 40 years of age. Males are affected more than females1. The use of molecular techniques like immunohistochemistry (IHC), polymerase chain reaction (PCR) and fluorescence in situ hybridization (FISH) is required for confirmation of diagnosis. They include detection of translocation T(x; 18) (p11.2; q11.2) is a highly specific gene mutation for synovial sarcoma4.
Classically, synovial sarcoma has a biphasic pattern and is composed of sheets of spindle cells and sharply segregated epithelial cells forming gland-like areas5. Diagnosis of monophasic synovial sarcoma, particularly at an unusual location like the mediastinum, is a challenge as it is often misdiagnosed as primary pulmonary spindle cell sarcoma/carcinoma or metastatic carcinoma6,7. Further work-up including clinical, histopathological and immunohistochemical findings are core when the diagnosis is uncertain and molecular testing is necessary8. This is particularly important for cases like ours from developing countries where financial and infrastructural constraints are major drawbacks.
Case
A 40-year-old female presented with complaints of cough, chest pain and shortness of breath with progressive weight loss for the past year. The past medical history was unremarkable. The radiological study x-ray (PA view, Figure 1) showed a large round opacity in the lower zone of the right lung with small nodules in the middle and lower zones of the left lung. Bronchoscopy revealed a right-sided growth involving the middle and lower pulmonary lobes. The computed tomography (CT) of the thorax (Figure 2) showed a well-defined mass measuring 8×7×6 cm on the right lung, infiltrating the diaphragm with small nodules of 1–2 cm in the left upper and lower lung lobes. The origin of mass from lung supstance or pleura could not be identified with certainty.
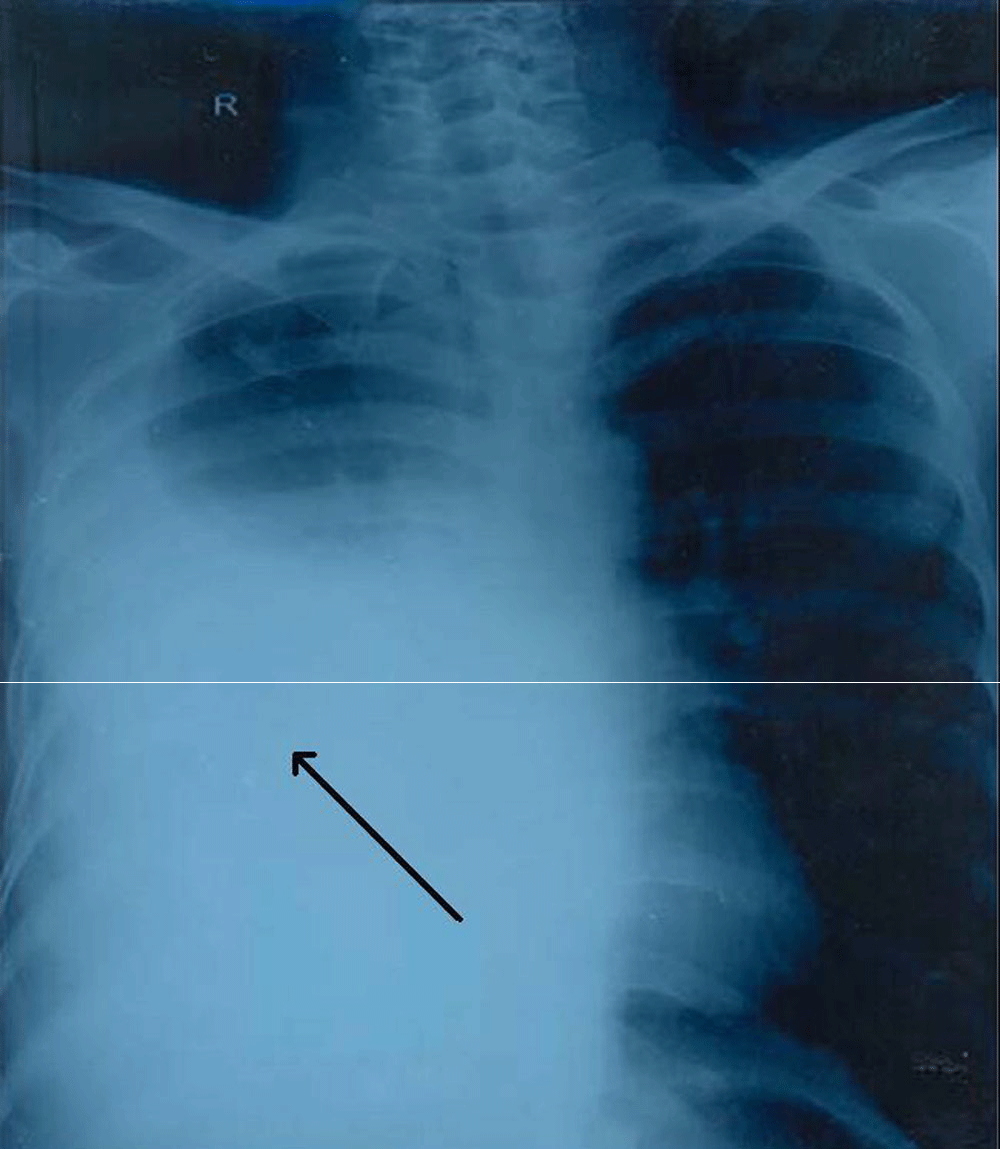
Figure 1. X-Ray Chest Arrow marked area shows a mass lesion on right side.
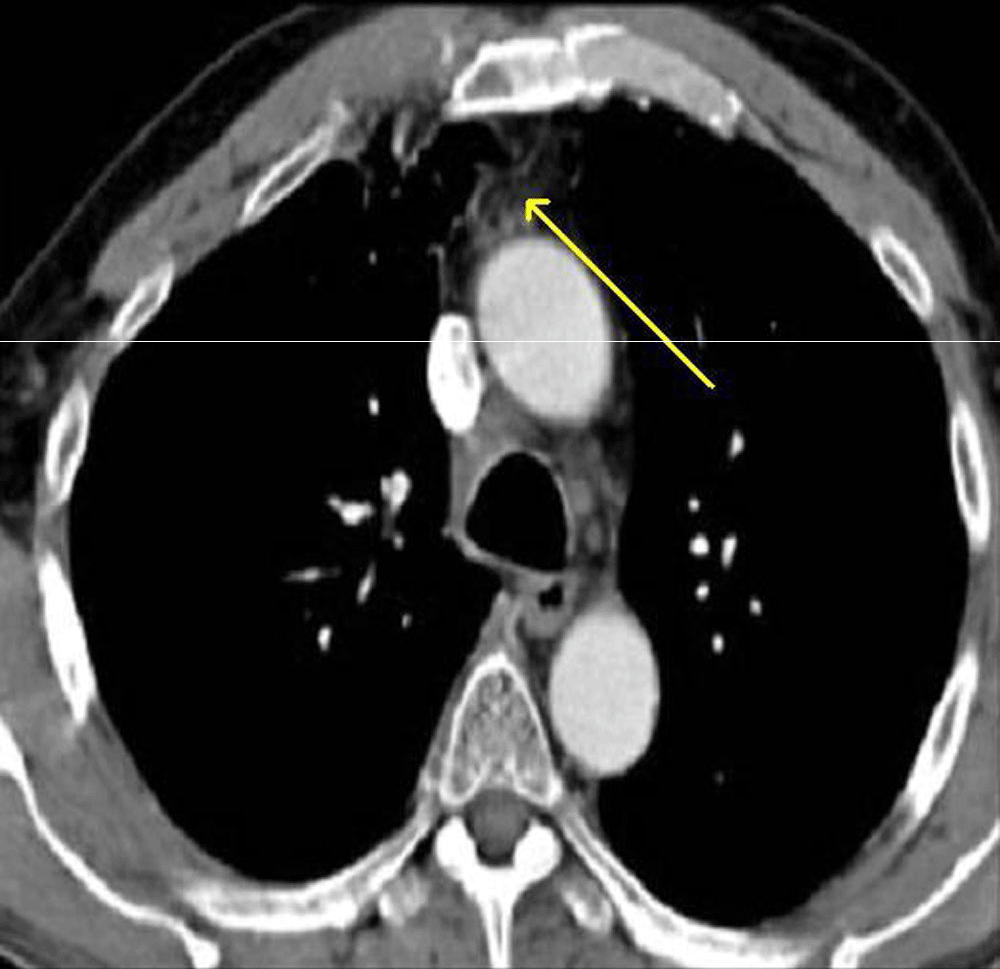
Figure 2. CT Chest Arrow shows tumor reaching upto pleura on right side.
CT-guided fine-needle aspiration cytology and bronchoalveolar lavage was inconclusive. Finally, a right side partial lobectomy was performed. On histopathology, we received three soft tissue pieces, the largest one measuring 19cm × 15cm × 10cm and consisting of part of the lung tissue with the tumor. A well circumscribed tumor measuring 19cm × 9cm × 9cm was seen. Figure 3 The cut surface of the tumor was fleshy, homogenous gray-white with areas of calcification. The other two pieces comprised of lung tissue measuring 7cm × 6cm × 2cm and tumor measuring 6cm × 6cm × 4cm.
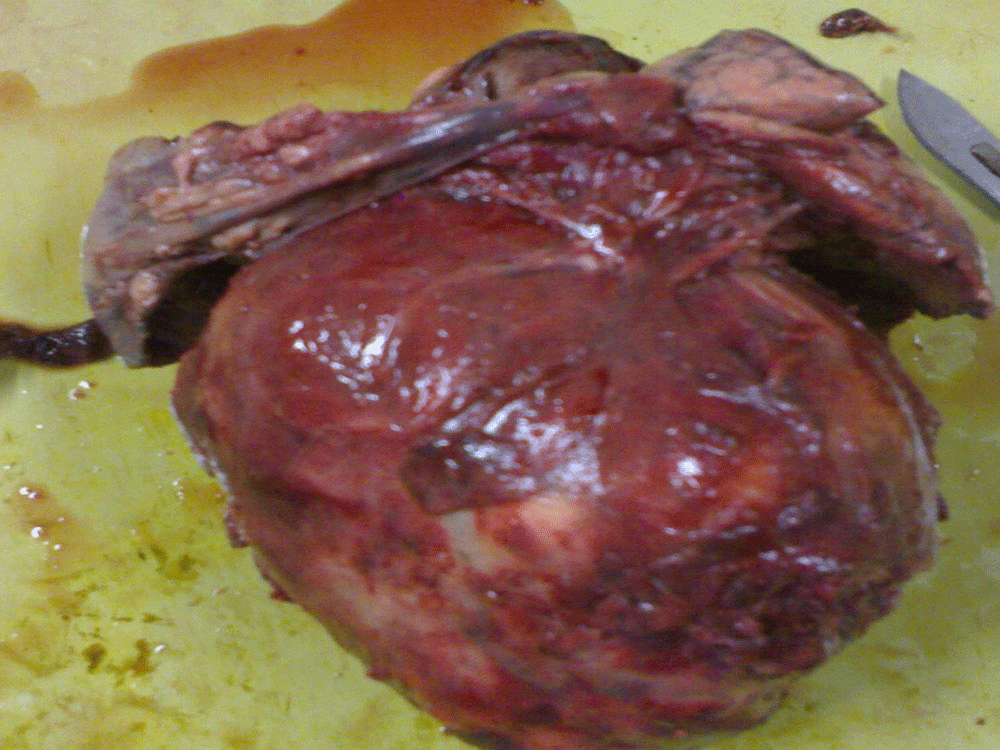
Figure 3. Gross photograph of excised tumor.
Microscopy examination showed a spindle cell tumor with a varied pattern of cellularity, with areas of high cellularity mixed with low cellularity areas (Figure 4 and Figure 5). The tumor cells were arranged in random, fasciculate and at places storiform pattern. Cytologically the cells had varying degrees of anaplasia with fusiform to plump cells having hyperchromatic nuclei and mild to moderate pink eosinophilic cytoplasm. Necrosis was minimal, there were occasional mitotic figures seen and metastatic calcification was also noted. It was therefore interpreted as a low-grade spindle-cell neoplasm of uncertain histogenesis. The tumor on immunohistochemical analysis was Epithelial membrane antigen cytokeratin5/6, mic-2, bci-2 and calponin positive. TTF-1, calretinin, WT-1 and CD-34 were negative. The tumor was therefore diagnosed as a synovial sarcoma.
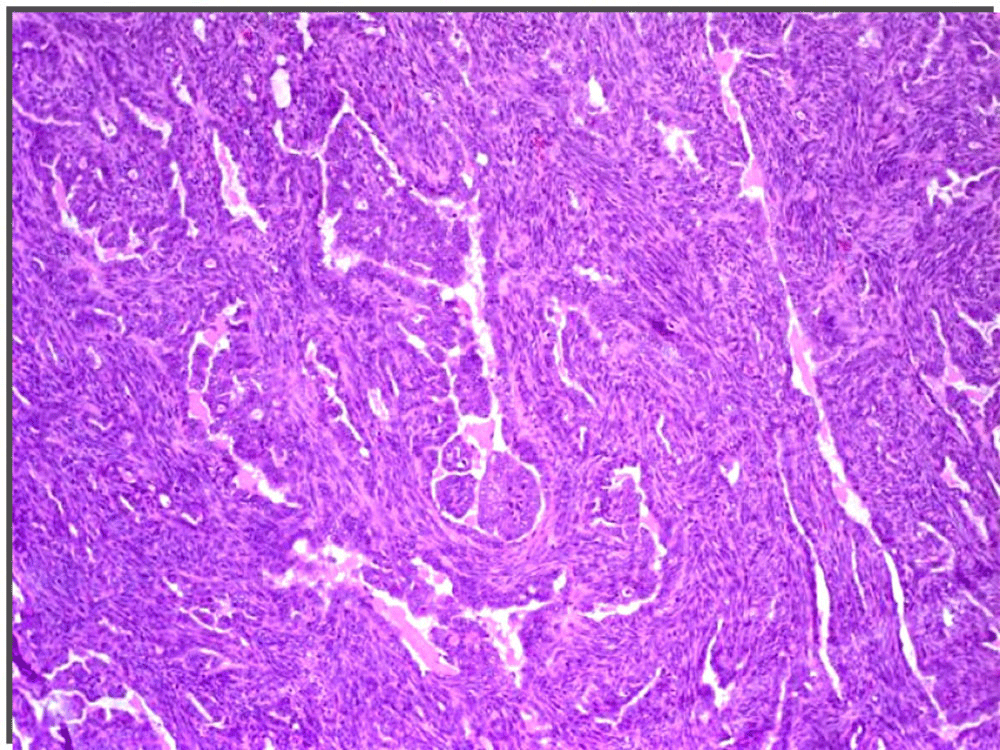
Figure 4. Microscopy: H& E 4x The spindle cell tumor cells arranged in random, fasciculate and at places storiform pattern.
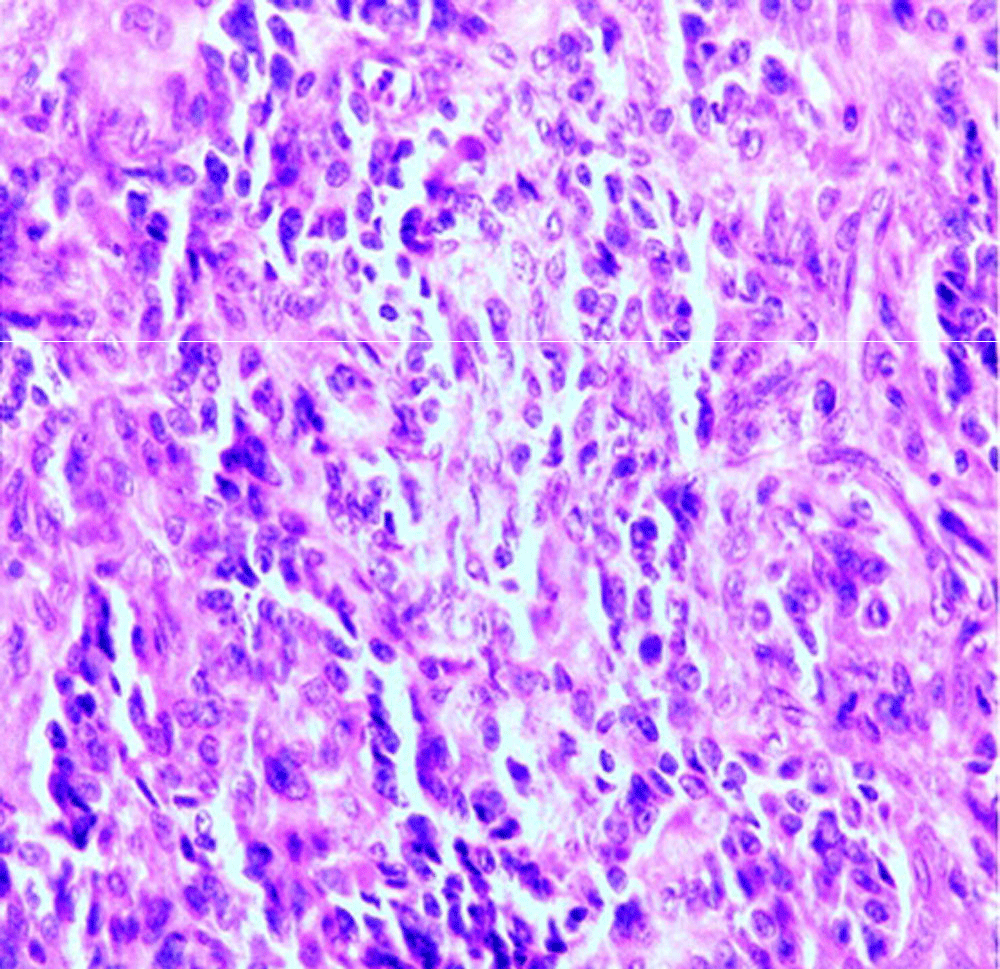
Figure 5. Microscopy: H& E 10x Shows plump cells having hyperchromatic nuclei and mild to moderate pink eosinophilic cytoplasm.
Discussion
The mediastinum contains various structures and pluripotent cells that are responsible for the origin of a variety of tumors at this anatomical site. As far as primary sarcoma is concerned, mediastinum sarcomas account for 1.4% of all soft tissue sarcomas. Among sarcomas of the mediastinum, the most common one is a malignant peripheral nerve sheath tumor (26%) followed by spindle cell sarcoma (15%), leiomyosarcoma (9%) and liposarcoma (9%). Synovial sarcoma accounts for 2% of all sarcomas of mediastinum9.
There are four morphological types of synovial sarcoma – biphasic tumor, monophasic tumor, monophasic epithelial tumor and poorly differentiated (round) tumor. The biphasic type is easily diagnosed due to the presence of epithelial and spindle cell components with sharply segregated epithelial cells forming gland-like areas2,5. Primary pulmonary monophasic synovial sarcoma is mainly of monophasic suptype and is difficult to diagnose due to its uniform spindle cell pattern. The differential diagnosis includes malignant peripheral nerve sheath tumor, fibrosarcoma, leiomyosarcoma, sarcomatoid variant of mesothelioma, Ewing’s sarcoma, hemagiopericytoma, spindle cell lymphoma, desmoplastic small round cell tumor and metastatic carcinoma6. Based on morphology, our first differential diagnosis was solitary fibrous tumor followed by mesothelioma. Ancillary immunohistochemical techniques helped to reach the final diagnosis. The immunohistochemical features of pleuropulmonary sarcoma are similar to those of soft tissue synovial sarcoma10. EMA, cytokeratin, and vimentin positivity in combination with CD-34 negativity are the most useful and sensitive biomarkers for diagnosis of monophasic synovial sarcoma. In our case, immunostaining was strongly positive for EMA, vimentin, bcl-2 and focally for cytokeratin. It was negative for thyroid transcription factor (TTF)-1, calretinin, WT-1, CD34, S-100 and CD-99. Theses markers ruled out the possibility of malignant peripheral nerve sheath tumor (S 100 Positive), primitive nerve sheath tumor (CD99 Positive), germ cell tumor (PLAP, Alpha AFP, HCG Positive) and leiomyosarcoma (SMA Positive). P63 and CD34 were negative, which further ruled out thymoma and solitary fibrous tumor. Bcl-2 was positive, but it was not specific, as it could be seen in solitary fibrous tumor and monophasic synovial sarcoma. The diagnosis of pleuropulmonary synovial sarcoma was based on an absence of tumor at any other primary site. Brain metastasis was not seen in our case as it is uncommon in soft tissue sarcoma with only a single reported brain metastasis in the literature11.
Table 1. IHC Profile of tumors10,12.
|
Tumor type
|
Cytokeratin
|
Vimentin
|
EMA
|
S-100
|
bcl-2
|
Calretinin
|
CD34
|
Desmin
|
|---|
| Synovial sarcoma | P | P | P | V | P | N | | |
| Mesothelioma | P | P | P | N | N | P | | |
| Leiomyosarcoma | | | | | N | | | P |
| MPNST | | | P | P | N | | | |
| Fibrosarcoma | P | V | V | N | | | | N |
| Angiosarcoma | | | | | | | P | |
The overall prognosis is poor in pleuropulmonary synovial sarcoma12. Tumor size more than 5 cm, male gender, advanced age >20 years, high mitotic activity >10 mitosis/10 HPF, presence of tumor necrosis and SYT-SSX1 gene variant are the main poor prognostic factors13.
To conclude, pleuropulmonary synovial sarcoma is a rare tumor. The diagnosis of this tumor requires meticulous clinical and pathological correlation, immunohistochemistry and molecular techniques. Familiarity with this entity is essential as it carries poor prognosis.
Comments on this article Comments (0)