Keywords
Adult onset Still’s disease, intracranial hypertension, papilledema, cerebrospinal fluid pleocytosis, IgE, IgG4, hemophagocytic lymphohistiocytosis
Adult onset Still’s disease, intracranial hypertension, papilledema, cerebrospinal fluid pleocytosis, IgE, IgG4, hemophagocytic lymphohistiocytosis
AOSD is an autoinflammatory, multisystemic disease that mainly presents in one of two distinctive clinical patterns, one with prominent systemic manifestations and another manifested predominantly with articular symptoms. These two patterns can overlap and either of these can be monocyclic (lasting from weeks to months), polycyclic, or chronic. Nervous system involvement has been reported in 7-12% of cases, manifesting with seizures, cranial nerve palsies, encephalitis, meningitis, stroke, demyelinating encephalopathy, peripheral neuropathy, and Miller Fisher-like syndrome.1-8
One of the most serious AOSD complications is HLH, also termed as macrophage activation syndrome in patients with autoimmune or autoinflammatory disorders. Approximately 14% of AOSD patients present that manifestation according to two retrospective studies including 176 patients, either at initial diagnosis or during follow-up, which significantly decreases their survival with a hazard ratio of 12.71, compared with non-HLH AOSD patients.9,10 Notably, AOSD was complicated by HLH in eight out of 20 patients in a French case series of AOSD patients requiring intensive care unit admission.11
On the other hand, intracranial hypertension can be associated with intracranial lesion, hydrocephalus, meningitis, encephalitis, encephalopathy, or with none of these. The rest of the cases are termed as idiopathic (pseudotumor cerebri) and can be caused by obstruction of cerebral venous drainage, medications and drugs used for other indications (e.g. insecticides) or other hematologic, neurological, endocrine or systemic disorders. Symptoms and signs attributed to intracranial hypertension can be bilateral papilledema, headache, vomiting, dizziness, abducens nerve palsy, tinnitus, visual or cognitive impairment (episodic or constant) and neck or back pain.
A 19-year-old Caucasian woman presented to the emergency department due to multiple febrile episodes the previous two weeks, along with loss of appetite and serious fatigue (she was able to walk only with assistance). Fever responded to paracetamol.
The patient was a computer engineering student. Her medical history, as well as her travel and sexual history were unremarkable. Her father had arterial hypertension and her mother had depression; no other significant information from the family history could be extracted. She denied special alimentary habits or close contact with people having fever. Some days ago, the patient had visited an Internal Medicine clinic where cefuroxime axetil 500 mg bid for 10 days was prescribed, but symptoms didn’t ameliorate. Blood tests had been ordered (Table 1), with the only notable result being the positive ANA test on a titer of 1/160, performed with enzyme-linked immunosorbent assay (ELISA). An upper abdominal ultrasound had been also performed showing splenomegaly, with maximum craniocaudal spleen diameter of 15 cm.
Vital signs on admission were as follows: 94 heart beats per minute, axillary temperature 39.2oC, blood pressure 110/70 mm Hg, oxygen saturation 100% and 24 breaths per minute while breathing ambient air and being totally alert. Heart auscultation revealed a mild systolic murmur, equally audible over the entire precordium. Searching for palpable lymph nodes revealed two non-tender, mobile right axillary nodes, sized approximately 1 cm2. Joint examination showed that all big joints were slightly warm with no other abnormal signs, while the patient was free of articular symptoms.
A 12-lead electrocardiogram, upright chest radiograph and urinalysis were normal. From a complete blood count, hypochromic and microcytic anemia was found along with low red blood cells, high erythrocyte sedimentation rate, and C-reactive protein (CRP) (Table 2). The findings above were considered as manifestations of anemia due to inflammation because they were accompanied by low serum iron and normal serum ferritin. A transthoracic cardiac ultrasound was ordered for heart murmur evaluation; it was totally normal and the murmur was subsequently attributed to hyperdynamic circulation in the context of febrile illness.
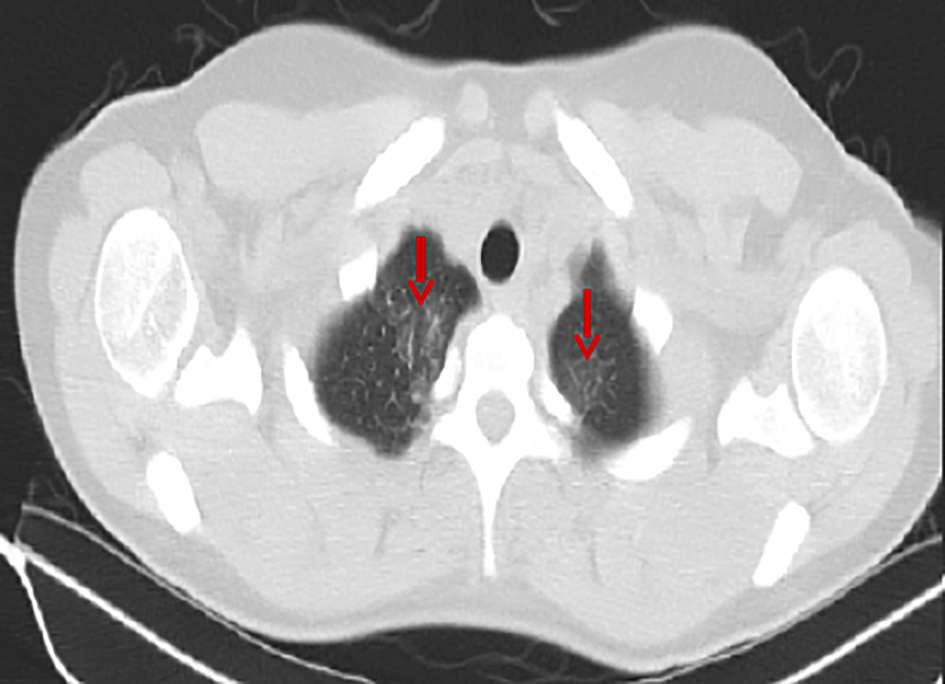
The 3rd hospital day fever continued and the patient developed thrombocytopenia and liver function test impairment (Table 2). The day after, she underwent an abdominal and chest computed tomography (CT), which revealed the already known splenomegaly and mild axillary lymphadenopathy, as well as bilateral pulmonary infiltrates, especially on the upper lung fields (Figure 1). With these findings doxycycline 100 mg bid and ceftriaxone 2 g qd were initiated with a working diagnosis of community-acquired pneumonia.
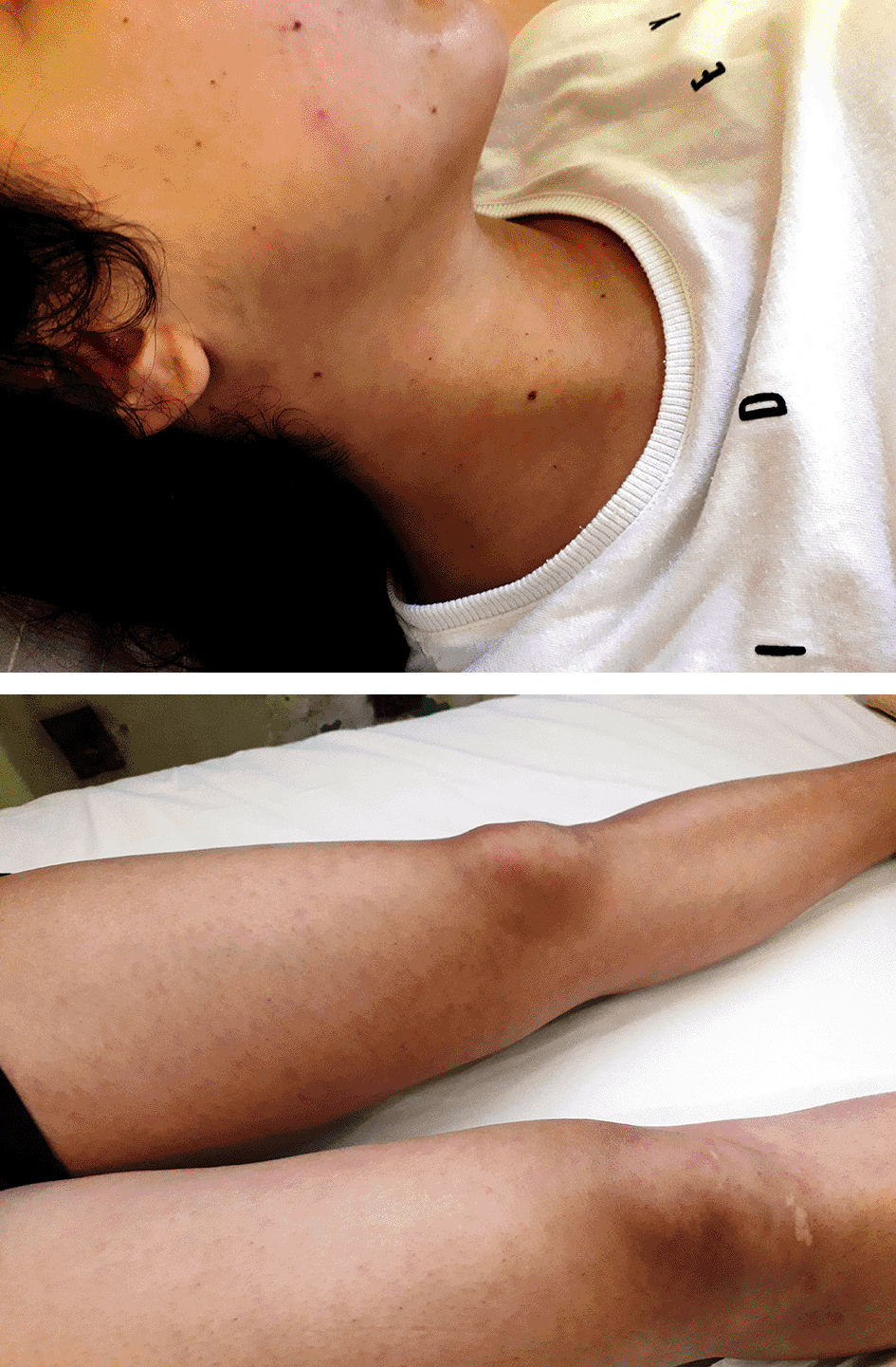
Approximately 24 hours after the first dose of antibiotics a mild, asymptomatic malar-papular reddish rash was discovered on clinical examination during a febrile episode and totally subsided when body temperature returned to normal range (Figures 2 and 3). This sign was also occurred in three other febrile episodes the next six days. Because fever persisted after four days of antibiotic treatment, despite a significant reduction of CRP values at the eighth hospital day (Table 2), the diagnostic workup was expanded to a thorough investigation for infectious and autoimmune diseases. All tests were normal, including ANA (performed twice with ELISA and indirect immunofluorescence by the hospital’s Laboratory of Immunology), except of an elevation of serum IgE and IgG4 immunoglobulins (Table 3).
Due to the aforementioned inconclusive results, the patient was scheduled to undergo a bronchoscopy in order to obtain a bronchoalveolar lavage for further evaluation of pulmonary infiltrates. Then, she had two episodes of vomiting without any concomitant symptoms or signs. Given the lack of any other obvious explanation, it was supposed to be a sign of intracranial hypertension. For that reason, an ophthalmologic consultation was obtained in order to perform a funduscopic examination. Bilateral papilledema was found; the rest of the examination was normal. With this finding the patient urgently underwent brain CT scan and brain CT venography in order to exclude venous sinus thrombosis, with no abnormal findings. A lumbar puncture was performed afterwards, revealing a very high CSF opening pressure (51 cm H2O), low CSF LDH (20 IU/L) and lymphocytic pleocytosis (Table 4).
Doxycycline was discontinued because tetracycline antibiotics can rarely elevate intracranial pressure, although more prolonged administration is usually a prerequisite.12,13 Ceftriaxone dosage was doubled to achieve better CSF concentrations and the following drug regimen was empirically added: IV ampicillin 2 g q4h for Listeria coverage, IV moxifloxacin 400 mg qd replacing doxyxycline against Staphylococcus aureus and atypical pathogens like Coxiella burnetii that can cause fever along with pulmonary infiltrates and CSF pleocytosis, IV ganciclovir for herpes virus family, IV dexamethasone 8 mg bid to cover immune-mediated disorders and acetazolamide tablets 250 mg qid, following the standard of care for idiopathic intracranial hypertension.
From the first day with that new regimen vomiting and fever subsided and the patient felt less fatigued. The next laboratory tests were improved except of a first appearing leucocytosis with neutrophil predominance, which was attributed to dexamethasone use (Table 2). CSF culture was sterile and multiplex CSF PCR (Table 4), as well as CSF cytology and serum West Nile virus antibodies were negative. For that reason blood PCR for Epstein–Barr virus, cytomegalovirus, human herpes virus 6 and 7, adenovirus and enterovirus was ordered, with also negative results.
In summary, the patient had rapid improvement after empirical treatment initiation, but a specific diagnosis was lacking. Because the workup for infectious causes was negative antimicrobial agents were ceased. Dexamethasone and acetazolamide dosage was decreased because of rapid improvement (4 mg bid and 250 mg tid respectively), with the patient continuing to be free of febrile episodes and in even better general condition. Meanwhile, the patient underwent a brain MRI with five days of delay until she got her dental braces off, with no abnormal findings.
In order to differentiate a self-limited viral infection from a non-infectious inflammatory process dexamethasone was discontinued. Two days after corticosteroid cessation fever reappeared and laboratory parameters deteriorated (Table 2). The aforementioned clinical course advocated that the diagnosis was an autoinflammatory or autoimmune disorder, with AOSD being first on the differential diagnosis because of the evanescent rash. Yamaguchi criteria were applied due to their highest sensitivity; the patient fulfilled six criteria, with two of them being major (fever above 39.2oC for >1 week and typical rash the two major criteria and enlarged lymph nodes, splenomegaly, abnormal liver function tests, negative ANA and rheumatoid factor the four minor), thus establishing AOSD diagnosis, which requires at least five criteria, including at least two major. Dexamethasone was subsequently reinitiated along with methotrexate 10 mg per week, folic acid 5 mg per week, calcium carbonate/cholecalcipherol 1.000 mg/800 iu fixed combination qd and trimethoprim/sulfamethoxazole (800 + 160) mg thrice weekly for Pneumocystis jirovecii prophylaxis. Influenza and Streptococcus pneumoniae vaccination was performed, too. Fever subsided again from the next day and CRP values declined (Table 2). Meanwhile, a gastrocnemius muscle biopsy was performed to exclude sarcoidosis more strongly based on studies from Andonopoulos et al and Yanardag et al; the biopsy was normal.14,15
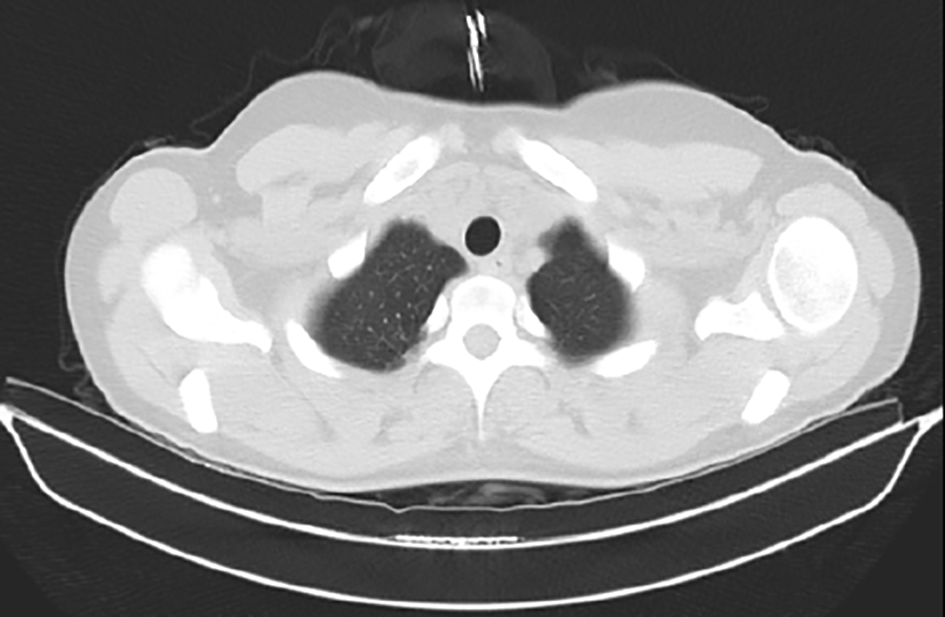
The patient was discharged and had been followed-up closely at the outpatient clinic. These visits included frequent funduscopic examinations from the department of ophthalmology. She remained asymptomatic and one month after diagnosis of intracranial hypertension papilledema had disappeared; acetazolamide was subsequently stopped. Papilledema or symptoms attributed to intracranial hypertension never presented thereafter. A repeated chest CT was performed 20 days after the first, showing complete resolution of pulmonary infiltrates (Figure 4).
The case of AOSD analyzed herein combined ordinary AOSD manifestations, such as evanescent rash and splenomegaly, with rare or unique manifestations. It should be emphasized that AOSD is a diagnosis of exclusion, even if the patient fulfills the applied criteria.
Pulmonary infiltrates is a relatively rare AOSD manifestation, usually resolving after successful treatment. It is found in 12.25% of the AOSD patients at the time of diagnosis according to a recent cohort from Italy.16 Besides that, the patient had normal white blood cell count and low platelets, with the exact opposite being a common finding in the majority of AOSD cases. That differentiation is usually found when AOSD gets complicated by reactive HLH, a serious and often detrimental complication. Further investigation of the patient included a bone marrow aspiration and biopsy, which revealed toxic bone marrow alterations reactive to systemic disease along with megakaryocytic and erythroid lineage suppression. Taking all parameters into account, HLH-score was calculated, resulting in a probability of 58% for HLH diagnosis.17 A high level of alertness for HLH recognition is needed, as it significantly increases mortality regardless of the precipitating disorder.
A classic AOSD regimen (corticosteroids plus methotrexate) was opted for without adding anakinra or other biologics because of rapid response, choosing dexamethasone over other corticosteroids because it was the drug of choice in many studies for HLH and the HLH-2004 protocol, too.18 Corticosteroids can be also used as monotherapy in AOSD, although an initial drug combination strategy is usually followed when risk factors for relapse after corticosteroid tapering exist like young age, splenomegaly, marked erythrocyte sedimentation rate elevation and very low glycosylated ferritin.
Concerning IgE elevation, its real prevalence and clinical significance is still unknown in AOSD. This finding has been anecdotally reported in the clinical course of AOSD, suggesting that it is either is pretty rare between AOSD patients or that IgE levels are not measured in a lot of AOSD cases, or both.19,20 Yokoi et al in their article had speculated that IgE levels in AOSD are dependent on disease activity.19
Elevated serum IgG4, on the other hand, has been primarily associated with IgG4-related disease, which is characterized by tissue infiltration from IgG4-producing B and plasma cells with concomitant serum IgG4 elevation in the majority of cases. However, recent data indicates that serum IgG4 levels may be also elevated in a variety of other diseases like helminthic infections, asthma and other eosinophilic disorders, primary sclerosing cholangitis, chronic hepatitis and liver cirrhosis, systemic vasculitides and other connective tissue diseases, cholangiocarcinoma, pancreatic cancer, lymphoma, plasma cell disorders, and multicentric Castleman disease, but has never been reported in AOSD before.21-24 Future research could better investigate this potential association.
The patient also presented some biochemical parameters found usually at IgG4-related disease except of merely having elevated serum IgG4. Specifically, IgG4/IgG ratio was above 0.114, element having great value for IgG4-related disease diagnosis according to Xia, Fan and Liu and IgG4/IgG1 ratio was above 0.24, a criterion used from Boonstra et al as a tool to discriminate IgG4-associated cholangitis from primary sclerosing cholangitis in patients with mild serum IgG4 elevation.23,24 However, IgG4-related disease was excluded based on clinical and laboratory grounds. These included high fever along with a rash and marked elevation of CRP values, the latter largely excluding IgG4-related disease.25 In addition, hypothesizing this was an IgG4-related disease case affecting central nervous system among others, the lack of characteristic pachymeningitis signs on brain MRI suggests an alternative diagnosis.
This case is the first AOSD case in the literature presenting intracranial hypertension with normal cerebral imaging and without neurological deficits or symptoms and signs of meningeal irritation. The possibility of idiopathic intracranial hypertension associated with doxycycline was excluded because of CSF findings. CSF pleocytosis in AOSD is neutrophilic in the majority of the cases, although lymphocytic pleocytosis has also been reported.6 We believe this case indicates that intracranial hypertension is a rare manifestation of AOSD in the context of subclinical meningitis. This is the reason why acetazolamide was added, extrapolating data from acetazolamide administration in cases of intracranial hypertension associated with systemic lupus erythematosus.26
Another interesting finding was the patient’s low CSF LDH levels (20 IU/L), as data from a 2009 study by Vázquez et al suggest that even in critically ill patients without brain trauma and decreased level of consciousness CSF LDH levels ≤40 IU/L are associated with a non-structural etiology of their symptoms (i.e., underlying diagnosis other than stroke, intracranial space-occupying lesion, encephalitis or meningitis).27 In addition, combining data from Quaglia et al and Lee et al who analyzed specimens from 157 patients with bacterial, viral or tuberculous meningitis, no sample had CSF LDH levels lower than 33 IU/L.28,29 Further studies should assess the prevalence and prognostic role of intracranial hypertension in adult-onset Still’s disease patients and if acute non-infectious, non-neoplastic meningitis is also accompanied with elevated CSF LDH.
The patient fulfilled the diagnostic criteria for AOSD. Treatment response to dexamethasone was dramatic, both initially and after the (almost immediate) recurrence of symptoms following the early trial to stop corticosteroids. This case report highlights that symptoms of intracranial hypertension may be the only ones when central nervous system is affected in AOSD. Even if acetazolamide was added to immunosuppressants, following dosing strategies for the management of idiopathic intracranial hypertension, it is not known if that medication contributed to positive outcome; future data should assess its role. Physicians taking care of patients with AOSD should be aware of that potential complication and thus promptly perform a funduscopic examination and a lumbar puncture if clinical suspicion exists.
Eleftheriotis G: Conceptualization, Methodology, Validation, Writing – Original Draft; Skopelitis E: Validation, Supervision, Writing – Review & Editing
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)