Keywords
Curcumin analogues, DYRK2, pharmacophore, docking, molecular dynamics simulation, ADME
This article is included in the Cheminformatics gateway.
Curcumin analogues, DYRK2, pharmacophore, docking, molecular dynamics simulation, ADME
Curcumin is a compound derived from turmeric (Curcuma longa), which is responsible for the yellow rhizome extract coloration. Traditionally, a large number of people in India, China, Indonesia and other Asian countries have applied turmeric powder in therapeutic herbs and as a food additive.1–5 The curcumin (diferuloylmethane) constituent is a tautomeric compound known to exist as an enolic form in organic solvents, and in keto form in water.6 The wide range of biological activities are currently being tested in vivo and in vitro to develop the numerous potentials in treating various diseases. These include the application of curcumin as an antioxidant, antibacterial, antifungal, antiviral, anti-inflammatory, and anti-angiogenic agent. Furthermore, there are reports on the promising anti-Alzheimer, as well as anticancer properties of curcumin, and its antagonistic effects against other degenerative diseases.7 The curcumin component is also a non-toxic compound, as no toxicity has been reported following the administration of high doses to animals.8 Previous reports have shown biological activities related to cancer, including lymphomas, breast, prostate, cervical, lung and colorectal cancers, alongside leukemia.1,4 There are numerous pathways involved in regulation, including p53, BAX, cyclin D1, various BCL, p21, p27, AKT, COX-2, protein kinase, and others.9
The facts and opinions on the biological effects of curcumin as a drug candidate indicate that it is a PAIN (pan-assay interference) compound or IMP (invalid metabolic panacea). Moreover, this unstable compound is known to easily degrade into others,10 and a total of eight have been reported to date. These include vanillin, ferulic acid,11,12 feruloyl methane, 2-hydroxy-6-(4-hydroxy-3-methoxyphenyl)-4-oxohexa-2,5-dienal,11 bicyclopentadione,13 ferulic aldehyde, vanillic acid,12 and 4-[(1e)-3-(propan-2-yloxy)prop-1-en-1-yl)guaiacol.14 In addition, numerous pharmacokinetic evaluations have indicated the poor absorption, low solubility, rapid metabolism and elimination as well as poor bioavailability properties of curcumin.15
There have been several suggestions of methods to solve these challenges, including co-administration with adjuvants. In addition, studies have shown the possible development into a nanoparticle form, complexations with metallic and radioactive elements, using the derivatives or analogue products, and application in the bioconjugation form.4,16 One of the current research strategies involves the screening of compounds from a large database to obtain analogues with the pharmacophore features of curcumin.
One of the purposes of studying curcumin and its analogues is to find analogues that are targeted to reduce cell proliferation by interacting with dual-specificity tyrosine-regulated kinase 2 (DYRK2). This is achieved through the positive regulation of the 26S proteasome, particularly in cancer cells. The inhibition property is observed in terms of cell proliferation with IC50 5 nM.17 DYRK2 is a family of protein kinases with members involved in cellular growth and development.18 In addition, there have also been reports on its function as a tumor suppressant by regulating cell survival, differentiation, proliferation and apoptosis.19 The mechanism adopted to control further involves the regulation of CDK14 expression.19 Furthermore, DYRK2 as an enzyme is capable of phosphorylating serine substrates and threonine residues. This action regulates apoptotic cell death in response to DNA damage by impacting the phosphorylation effect of Ser46 on p53.19,20 Furthermore, reports have shown the negative regulatory impact on breast cancer formation through the transcriptional downregulation of Kruppel-like factor 4 (KLF4).20
Therefore, the aim of this study is to explore the curcumin analogues for their potential application as a DYRK2 inhibitor through virtual screening by using pharmacophore molecular modelling as well as docking and molecular dynamics. The compounds of screening results are expected to be applied as lead compounds in discovering and developing a prospective anticancer molecule through DYRK2 inhibition.
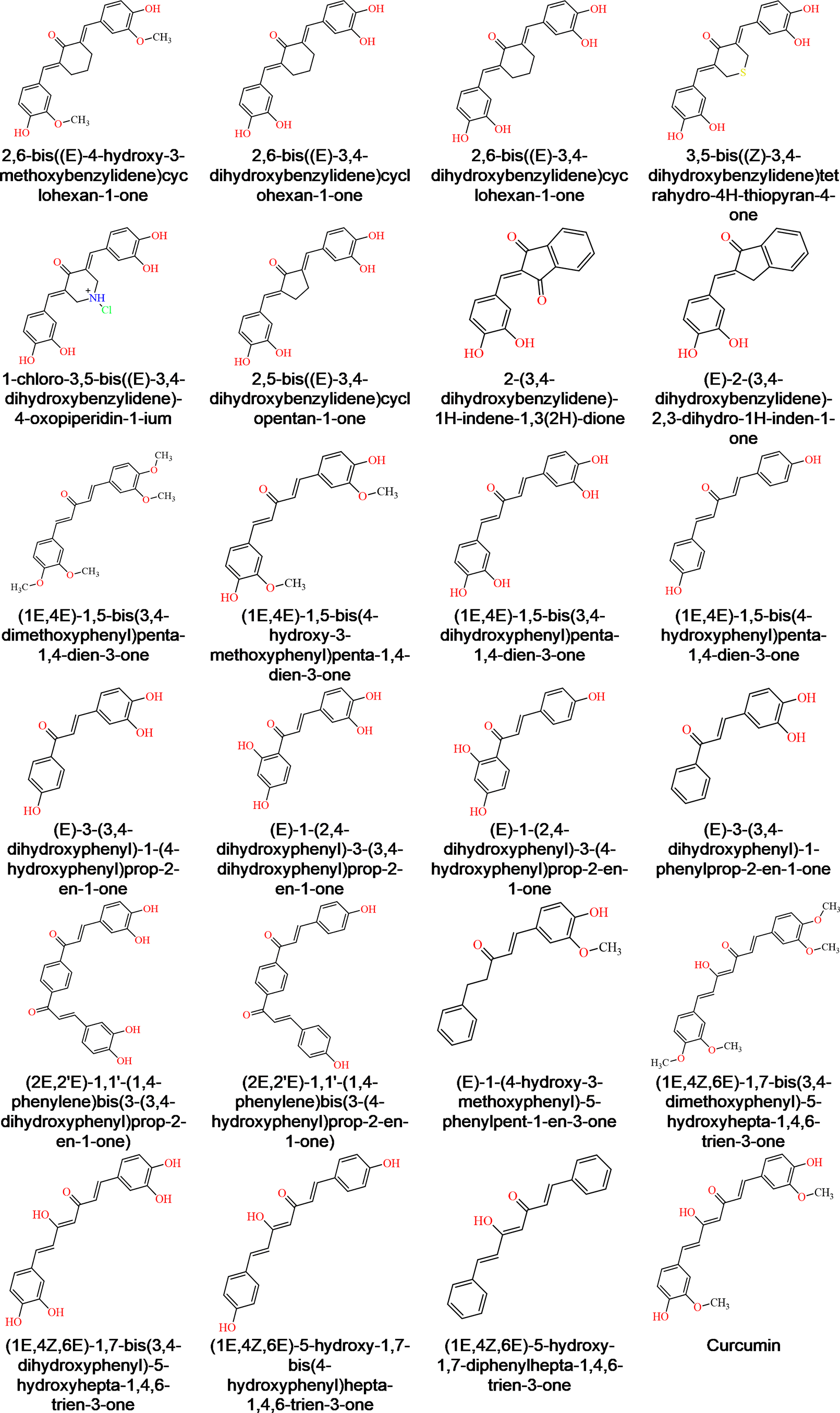
Figure 1 shows the chemical structure of curcumin and the analogues21 used to model the dataset for ligand-based pharmacophore. The 2D chemical configuration was constructed with MarvinSketch 19.2, prior to an analysis with LigandScout 4.3 win64 evaluation version22 (the analysis can be replicated using PharmaGist Webserver). Subsequently, each structure’s geometry was optimized using the energy minimize module with MMFF9423 force field set at a default setting.
5ZTN was the Protein Data Bank accession number of the DYRK2 protein used in this study,17 and curcumin acted as the native ligand. In addition, the target for molecular docking and dynamics was prepared using Molecular Operating Environment (MOE) 2014.0901 software (this can be replicated using MGLTools 1.5.6 and USCF Chimera 1.13.1) in order to correct the break residues, charging, and protonation of the protein structure. The protein molecule was opened in AutoDockTool (ADT) 1.5.6, and the water molecule(s) were then removed. In grid menu, macromolecule was chosen, and the protein structure was then saved as a.pdbqt extension.
The ligand-based pharmacophore observed in this work was analyzed through multiple flexible alignment. Therefore, the model was generated from 24 dataset compounds using LigandScout 4.3 software (or PharmaGist Webserver). This was achieved through the 3D superposition of chemical features constructed by the flexible conformation alignments of all dataset compounds. In addition, the enrichment factor (EF) and receiver operating characteristics (ROC) analysis were used to validate the pharmacophore model using ROC Analysis: Web-based Calculator for ROC Curves. Active compounds were all of the 24 dataset compounds, and decoy compounds were obtained from zinc decoy database generated via DecoyFinder 2.0.
MolPort provided a large database with over 7.4 million catalogue compounds. The process of curcumin analogue filtration from the compounds database was performed on the Pharmit webserver. Filtering the compounds from database was then conducted using the pharmacophore query file as obtained from the above pharmacophore modeling.
The goal of molecular docking was to assess the binding affinity of compound(s) upon interaction with the receptor. Therefore, all results obtained from the database filtering process were docked to the DYRK2 protein. In addition, the docking module of MOE was used for docking protocol detection and also for the docking score calculation of all hits (this can be replicated using AutoDock 4.2.6). Moreover, the molecular docking protocol was evaluated through virtual screening with the alpha triangle methods, London dG scoring and GridMin refinement.
The aim of molecular dynamics was to evaluate the physical movement of molecules and atoms. This activity was intended to stimulate the interaction stability between the ligand and DYRK2, and was further investigated in combination with protein-ligand complexes obtained from the docking score calculation and characterized by the highest binding affinity. In addition, the interaction dynamics between ligands and receptors was measured using Gromacs 2018.3.24–29 The stability of ligands in the binding pocket of DYRK2 protein were simulated by the molecular dynamic for 50 ns. In addition, Gormos96 54a7 force field was used to prepare the protein topology, while the PRODRG webserver was applied for ligand topology, using Gromos forcefield. The complex protein-ligand was solvated in a dodecahedron with 1 nm dimensions. Moreover, an aqueous environment was created in the system with the simple point charge (SPC) water model, and this was neutralized by adding Na+ and Cl–. The electrostatic interaction and periodic boundaries were calculated in all conditions using Particle-mesh Edward (PME) methods. Meanwhile, the cut-off radius for short-range van der Waals and Coulomb interactions was set to 0.9 nm. Furthermore, the linear constraint solver for molecular simulations (LINCS) method was used to constraint all bond lengths, while minimization, NPT and NVT equilibration as well as system production were performed at constant temperature (300K) and pressure (1 atm). The minimization process was conducted for 50 ps, NPT and NVT were collectively simulated for 100 ps, while the production process for 50 ns were saved at every 2 ps with coordinates of each simulation.
The interaction of ligand-receptor was visualized with LigandScout 4.3 (and can be replicated in USCF Chimera 1.13.130 and Discovery Studio Visualiser v20. The energy from ligand-receptor interactions were further estimated using the g-mmpbsa31 platform.
The absorption, distribution, metabolism and excretion (ADME) prediction values for hit compounds were calculated using ADMET Prediction by ADMETLab Webserver. Furthermore, ADME properties applied in the current research include caco-2 permeability, bioavailability 30% (F30), plasma protein binding (PPB), blood-brain barrier (BBB), Cyp450 1A2 inhibitor, Cyp450 1A2 substrate, half-life (T1/2) and clearance (CL).
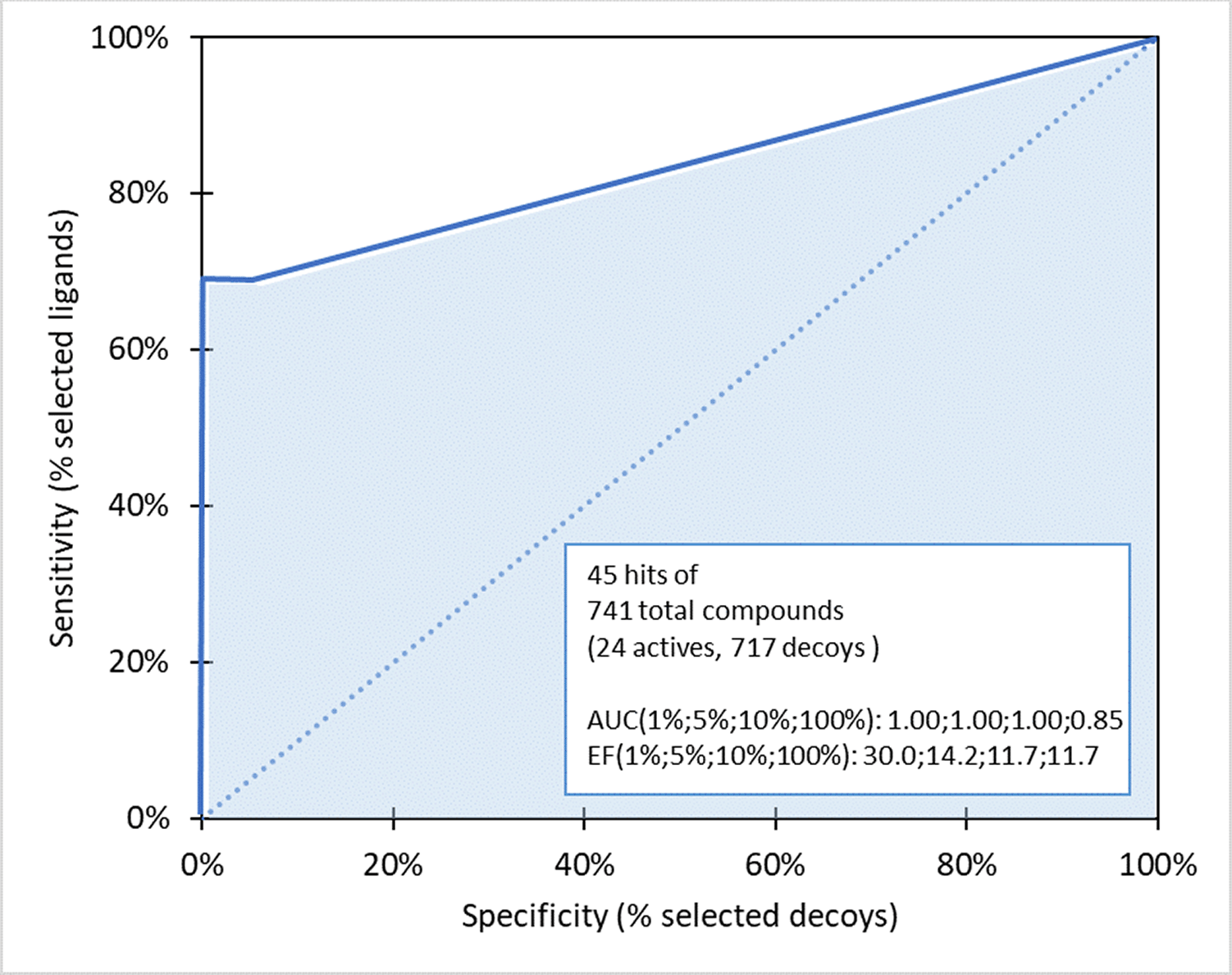
The hypothetic pharmacophore was grouped based on the number of features, comprising 3 to 7, and each has 10 models, totaling to 50. Figure 1 shows the structure of the dataset molecules used to construct the pharmacophore model, where the validation process including ROC and EF analysis were implemented. These were performed on 24 active compounds and 717 decoy compounds obtained from zinc decoy database generated by DecoyFinder 2.0. Table 1 summarizes the values of area under the curve (AUC), ROC curve and EF for all models.
Table 1 shows the adequacy of AUC values for all models, where the highest EF value of was observed in model4-10 and was consequently selected as the best model. Figure 2 shows the ROC curve indicating the composition of 45 hit compounds, including 17 active and 28 decoy. Meanwhile, AUC values shown on 1%, 5%, 10% and 100% were 1.00, 1.00, 1.00 and 0.85, respectively, while the corresponding EF values were 30.8, 14.2, 11.7, and 11.7.
The output file of the selected pharmacophore model generated from LigandScout 4.3 (or PharmaGist Webserver) was used to filter the compound database. Furthermore, the Pharmit interface was used for 3D visualization of the features, including details on the coordinate position and radius. The presence of hydrophobic, H-bond acceptor and donor features with radius 1.5 Å, and 0.9 Å for the aromatic variant were observed in the default setting. Moreover, filtering on Pharmit allowed the users to modify the feature’s radius, and consequently increase or decrease the amount of hit(s) as a result. However, manually changing this value is also possible by modifying the hit reduction and screening criteria. The Pharmit developers enter some criteria to set up the maximum hits per conformation, and molecule number, as well as the total limit for reduction, molar weight, rotated bond, logP, polar surface area (PSA), aromatic ring, H-bond donor and acceptor for screening.
This novel work involves database filtering with default settings for pharmacophore features and hit screening, with the exception of reduction. The model ultimately produced 1,130 hits, and a lot more compounds exist for continued screening. Therefore, the feature radius and screening procedure were modified. In particular, the hydrophobic, H-bond acceptor and donor radius were reduced to 1.4 Å for each, while the Rule of Three (RO3)32 was applied during hit screening. These rules include molar weight (300 Dalton), rotated bond (3), logP (3), aromatic ring (3), H-bond donor (3) and H-bond acceptor (3). Meanwhile, PSA was set to the maximum value according to another reference, at 90 Å,33 and these filtering protocols collectively produced 566 hits.
Groups of model3, model4, model5, model6, and model7 have 3, 4, 5, 6 and 7 chemical features, respectively. Every group has 10 model candidates. AUC, area under the curve; EF, enrichment factor.
Binding affinity is an indicator of the connection strength between ligand and receptor. This was determined by a docking score calculated using the MOE software (and can be replicated using AutoDock 4.2.6). This procedure was then validated by redocking the native ligand of DYRK2 protein (5ZTN) present in curcumin. The lowest RMSD value for successful docking was 0.7788, indicating the propensity to apply this protocol to other ligands.
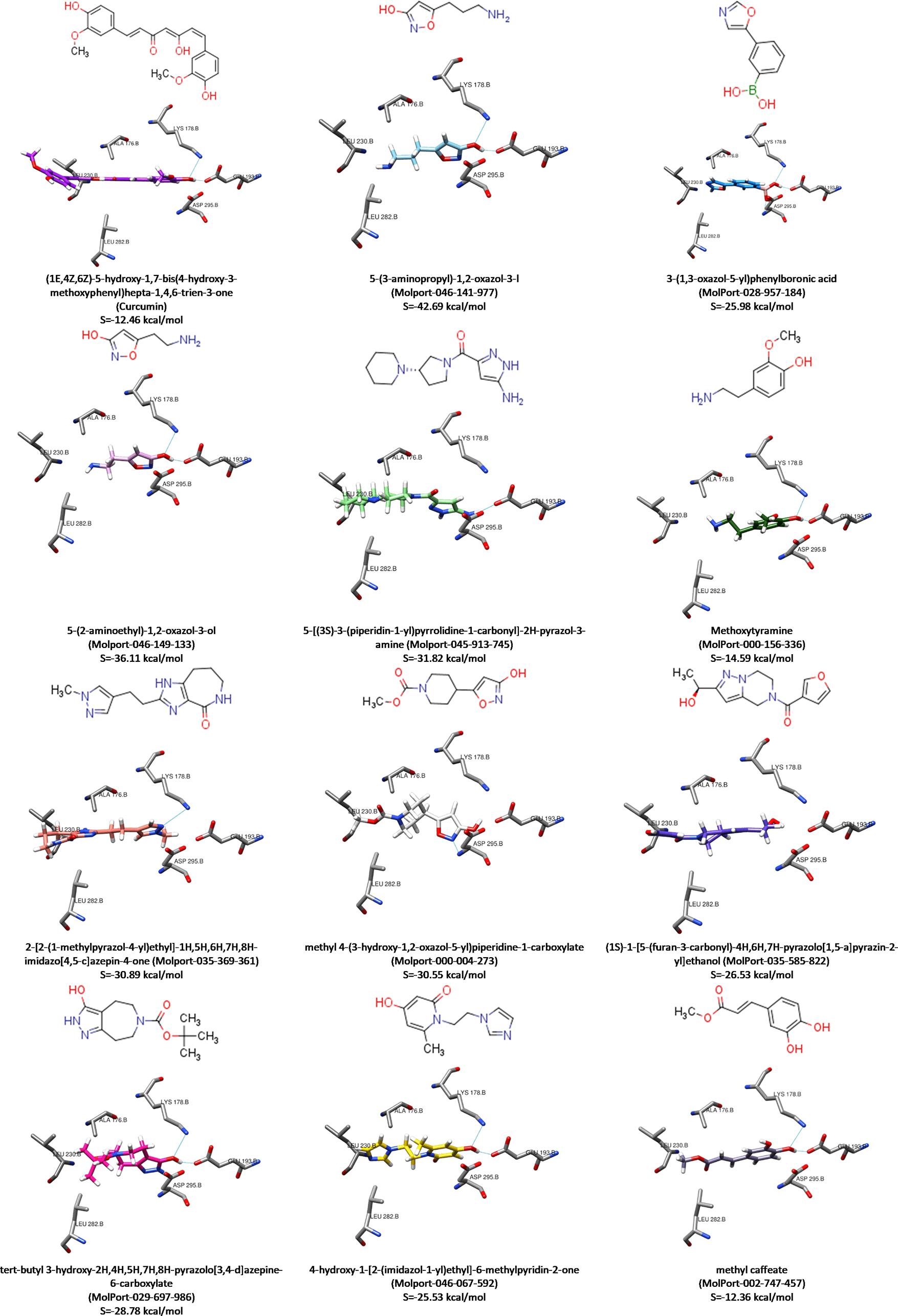
This protocol docking validation process involved calculating the binding affinity (docking score) for each curcumin analogue complex and DYRK2 protein. Moreover, the value obtained for curcumin during redocking was –12.46 kcal/mol. This was used to filter the hits, as the specimens with greater and relatively close values between the native ligand and the binding pocket of target were selected. Figure 3 shows the chemical structure of ligand (hit compounds) and ligand-DYRK2 interaction with docking score lower than –10 kcal/mol, and Figure 4 indicates the overlay of each, with pose characterized by the highest value.
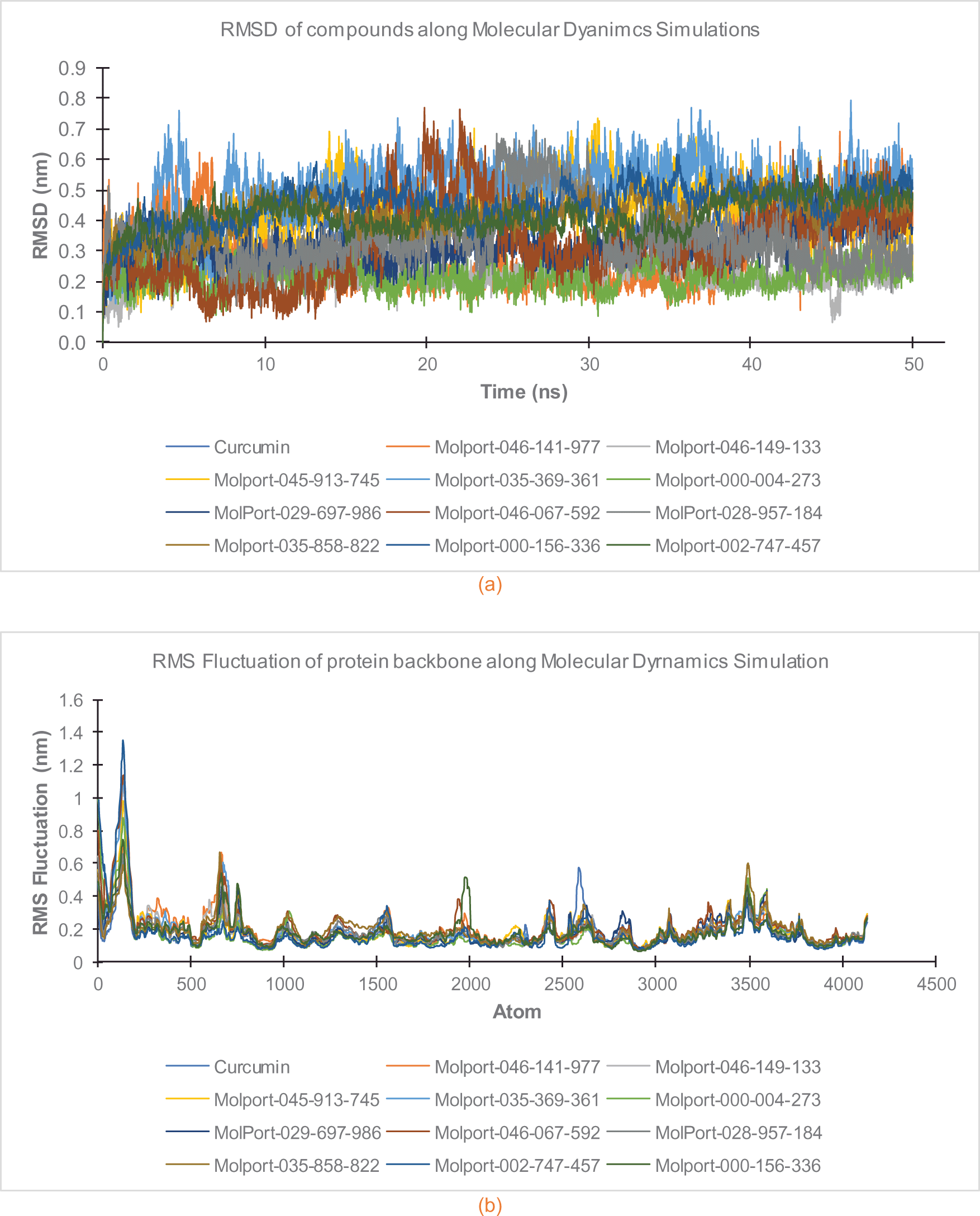
The complexes (ligand-protein) motion during simulation were expressed in Root Mean Square Deviation (RMSD) (Figure 4(a)), while the movements of protein backbone during simulation was estimated in a Root Mean Square Fluctuation (RMSF) curve (Figure 4(b)). Figure 4(a) showed that the interactions are stable after 30 ns of simulation.
The ADME (absorption, distribution, metabolism, and excretion) provide a description for drug disposition within an organism. These pharmacokinetic parameters influence the overall level and kinetics in the tissues, and consequently influence the pharmacological effect of the active compounds. In addition, it is possible to represent the ADME of active compounds as a prediction value.
The results obtained with the selected compounds in this current investigation were caco-2 permeability and F30 for absorption; PPB and BBB for distribution, Cyp450 1A2 inhibitor and Cyp450 1A2 substrate for metabolism; while T1/2 and CL represented excretion. Table 2 shows the summary of ADME prediction for selected compounds in contrast with curcumin.
F30, 30% bioavailability; PPB, plasma protein binding; BBB, blood-brain barrier, T1/2, half-life CL, clearance.
The pharmacophore is the physicochemical feature of a molecule known to interact with a specific target receptor. This is modelled in a 3D pattern and the basic characteristics are also shared by a set of molecules. The ligand-based pharmacophore model was constructed without needing a protein target structure, through a 3D superposition of the ligand conformed physicochemical features.
The chemical characteristics of a selected pharmacophore model include three H-bond acceptors provided by the oxygen in keto, methoxy and hydroxyl groups, a H-bond donor from the hydroxyl, an aromatic feature and hydrophobicity contributed by aromatic group. Figure 5(a) shows the 2D visualization of pharmacophore model, while Figure 5(b) and 5(c) demonstrates it in 3D.
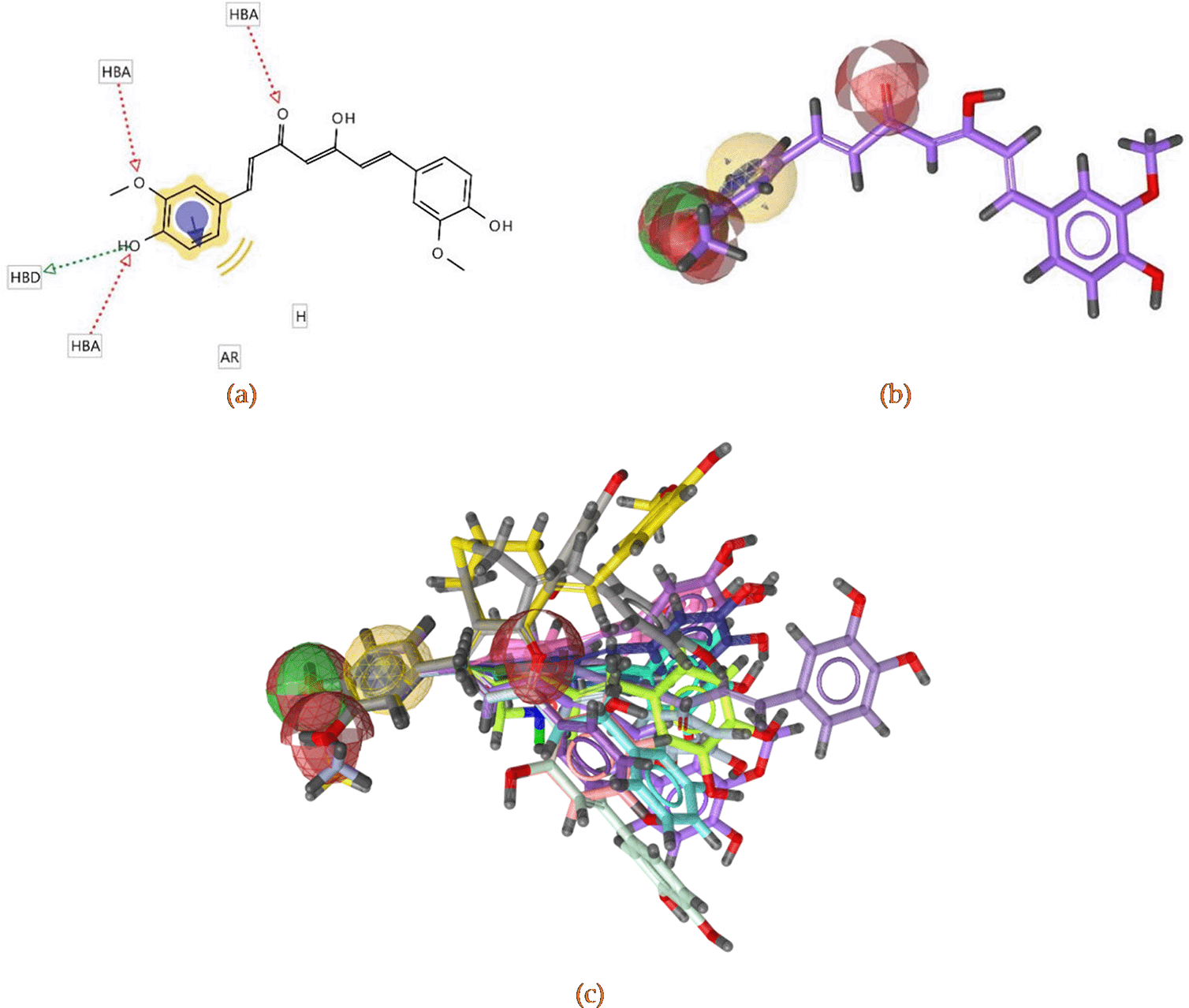
3D visualization of pharmacophore model (red balls are H-bond acceptors, the green are H-bond donors and the yellow are aromatic and hydrophobic features) (b). Alignment of 17 hits of active compounds in the pharmacophore features (c).
There is rule of three (RO3) beside Lipinski’s rule of five (RO5), which applies in searches pertaining to drug-likeness. Particularly, RO5 stipulates the criteria to be satisfied, including the presence of no more than five hydrogen bond donors, 10 hydrogen bond acceptors, a molecular weight (MW) of less than 500 Daltons, and an octanol-water partition coefficient (logP) below 5.34,35 Furthermore, all numbers are in the multiple of five, hence the RO3 criteria requires a multiple of three. These criteria include logP not greater than 3, MW less than 300 Daltons, the presence of no more than three hydrogen bond donors and acceptors, respectively, and no more than three rotatable bonds.36
The Pharmit platform allows users to customize the pharmacophore feature criteria based on RO5 or RO3. In addition, the value modification potentially increases or decreases the amount of hits. The application of RO3 in this research instigated a decline in the number of hit compounds from 1,130 (default criteria) to 566 (modified criteria). The latter was determined to be more rational during molecule screening.
Molecular docking is one of the in silico approaches of interaction studies between ligand(s) and receptor. The 566 hits are further filtered through this means, and 11 compounds were selected after using the specified protocol. In addition, the binding affinity (docking score) of samples in each hit were better than or close to curcumin (native ligand). Figure 6 shows the respective overlay with the binding pocket of DYRK2 protein. It was observed that all selected hit compounds occupy the DYRK2’s binding site.
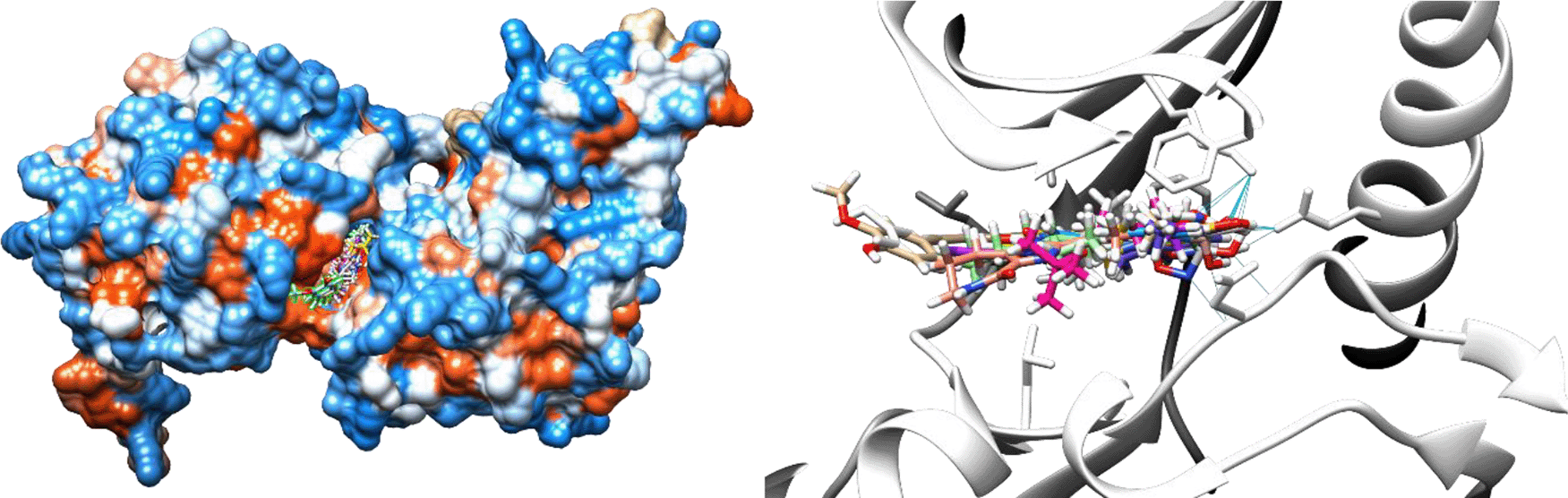
Figure 4(a, b) shows the stability performance of RMSD for each ligand, as well as the RMSF for the respective protein backbone on each complex, after the initial 25 ns simulation. Therefore, an average of the system binding energy was calculated at the end. The van der Waals, electrostatic, polar solvation, non-polar (Solvent-Accessible Surface Area) and binding energy were calculated every 200 ps, in order to obtain an average value for each complex. Table 3 shows the summarized system energy report obtained from 200 snapshots.
The native ligand (curcumin) has a binding energy of –53.058 kJ/mol with the DYRK2 protein. Table 3 showed six selected hit compounds have more negative values compared to that of Curcumin, i.e. Molport-035-369-361 (–71.35±24.85), Molport-000-004-273 (–83.56±18.24), MolPort-029-697-986 (–61.69±14.89), MolPort-035-585-822 (–74.24±15.55), MolPort-000-156-336 (–54.49± 15.09), and MolPort-002-747-457 (–69.36±12.58).
This shows that van der Waals and polar solvation energy are the main impact on binding factors for curcumin and all selected compounds. The respective values were negative and positive. This indicates the tendency to generate more positive effect on binding energy at more negative van der Waals. The binding energy of all six selected compounds were larger (more negative) than that of curcumin. This discrepancy was attributed to the lesser polar solvation characteristics. Moreover, significantly higher values of positive polar solvation energy tend to decrease binding energy.
Figure 7 showed the position of curcumin and other selected hit compounds in the binding pocket of DYRK2 during the molecular dynamic simulation. These results confirm those of our docking study. The interaction of 5ZTN-curcumin (Figure 8(a)) and 5ZTN-Molport 000-004-273 (Figure 8(b)) show the binding mode which involves H-bonds and hydrophobic interactions.
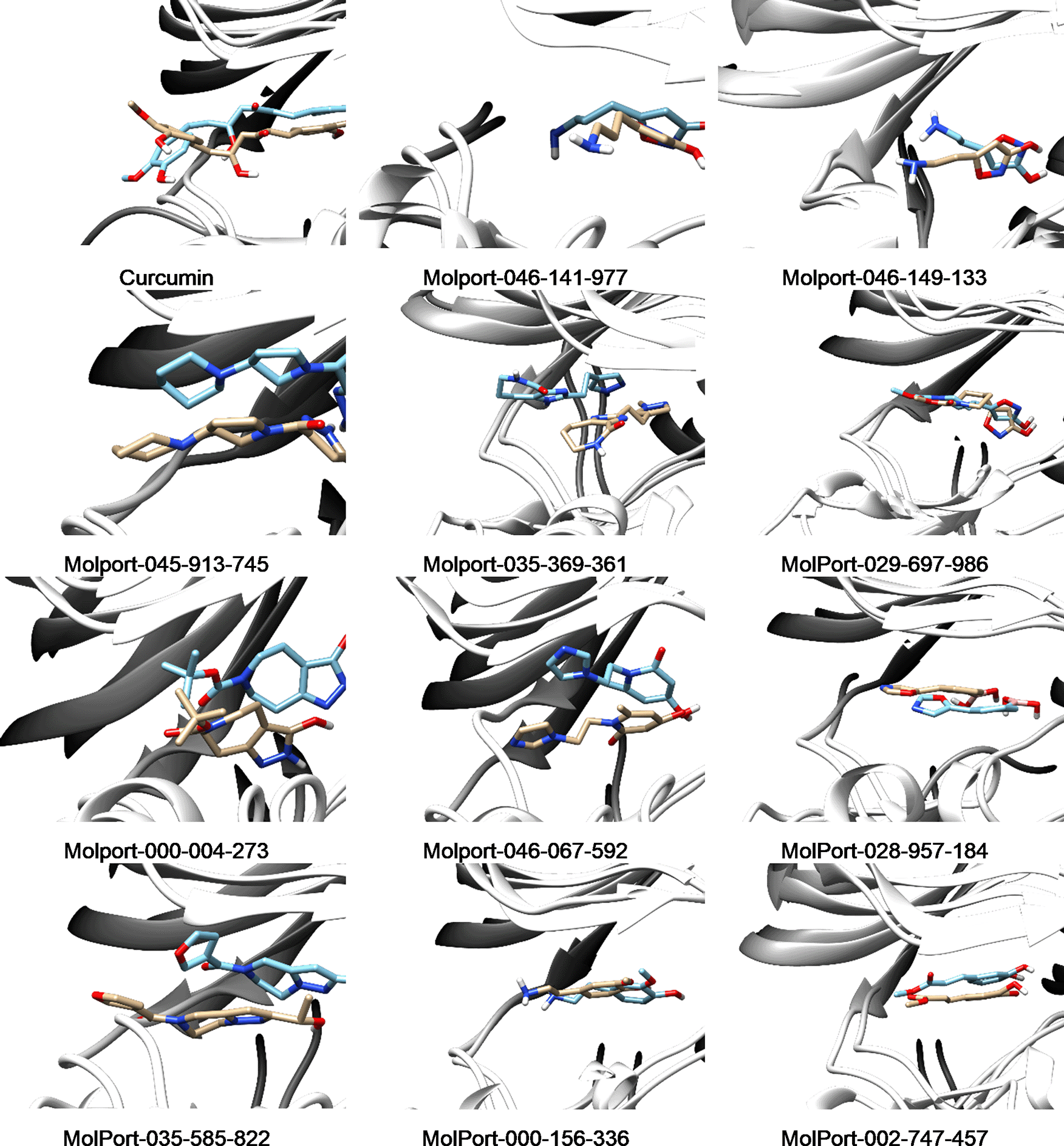
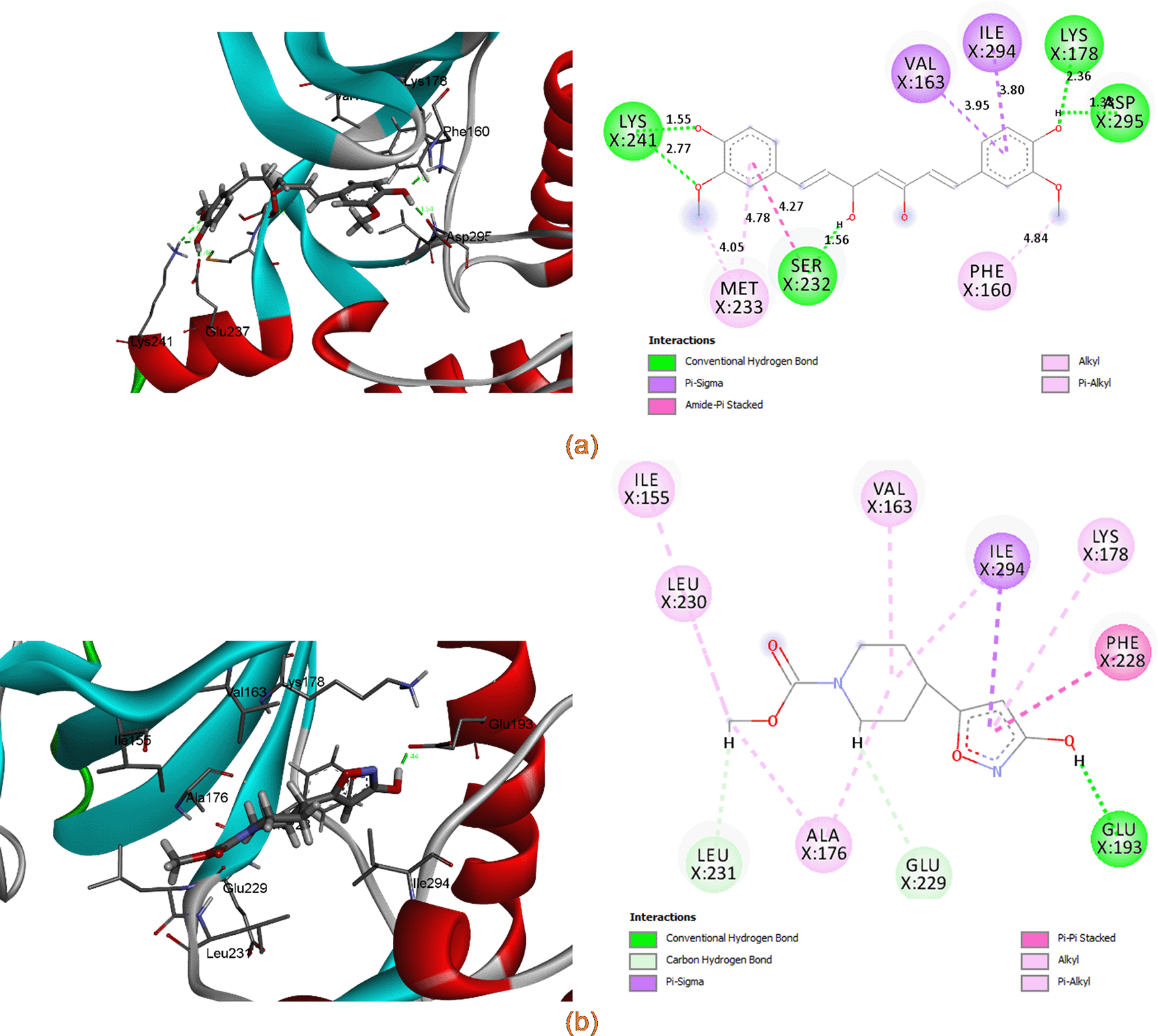
Table 4 summarizes the percentage of H-bond occupancy between the selected hit compounds and residues of DYRK2. It was observed that LYS178, GLU193, ASP295 have important roles in the ligand interaction in the DYRK2 binding pocket.
The absorption prediction results recognized all 11 selected compounds to be better than curcumin in terms of Caco-2 permeability, where the optimal output for active compound is suggested to be more than –5.15 log cm/s.37 The calculated values were above –5.0, whereas a value of –5.13 was recorded for curcumin. In addition, the bioavailability (F30) was also better, as the curcumin was categorized 0 (<30), while the selected compounds were attributed as 1 (>30%) except MolPort-029-697-986 or tert-butyl-3-hydroxy-2H,4H,5H,7H,8H-pyrazolo[3,4-d] azepine-6-carboxylate.38
The PPB was higher in curcumin, at 88.84% compared to the others. Hence, the native ligand is considered to have a more significant bond with plasma protein, although less than the suggested 90%, and consequently has a lower therapeutic index.39 Conversely, the selected compounds have a greater propensity to attach to the target receptors and provide the desired therapeutic effect.
The BBB is a highly selective semipermeable border separating the circulating blood from the brain and other extracellular fluids in the central nervous system (CNS).40 This parameter is calculated as a ratio of compounds, and is improved by H-bond numbers as well as molecular weight.41 The ADMETLab webserver categorizes the BBB value of a drug as 0 (BBB negative) indicating the inability to penetrate, whereas 1 (BBB positive) demonstrates a barrier permeation potential. In addition, all compounds, including the curcumin were categorized with a BBB value of 1, and a positive BBB is derived in instances where the ratio probability was above 0.1.
The cytochrome P450 (CYP) gene family are responsible for drug metabolism. Here, the ADMETLab webserver was used to predict the interaction potentials of curcumin and all selected compounds as an inhibitor and substrate of the cytochrome P450 enzyme. However, the drugs were unable to act as inhibitors but could act as a substrate of cytochrome P450. The prediction results showed the tendency for 2-[2-(1-methylpyrazol-4-yl)ethyl]-1H,5H,6H,7H,8H-imidazo[4,5-c]azepin-4-one (Molport-035-369-361), (1S)-1-[5-(furan-3-carbonyl)-4H,6H,7H-pyrazolo[1,5-a]pyrazin-2-yl] ethanol (MolPort-035-585-822), and methoxytyramine (MolPort-000-156-336) to act as a substrate and not as CYP1A2 inhibitor. Furthermore, 4-hydroxy-1-[2-(imidazol-1-yl)ethyl]-6-methylpyridin-2-one (Molport-046-067-592), and 3-(1,3-oxazol-5-yl) phenylboronic acid (MolPort-028-957-184) potentially act as substrate and inhibitor. Curcumin or (1E,4Z,6Z)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one, Methyl 4-(3-hydroxy-1,2-oxazol-5-yl)piperidine-1-carboxylate (Molport-000-004-273), and tert-butyl 3-hydroxy-2H,4H,5H,7H,8H-pyrazolo[3,4-d]azepine-6-carboxylate (MolPort-029-697-986) were predicted as being unable to act as inhibitor and a substrate of cytochrome. The three other compounds can only act as a cytochrome inhibitor but cannot act as a substrate.
CL refers to the volume of plasma of the drug freed per unit time, where (T1/2) is half-life in plasma. The results indicate lower values with hits and curcumin at < 5 mL/min/kg and < 3 hours, respectively.39 Curcumin possessed the comparable CL and the highest T1/2. Thus, selected hit compounds are considered to have relatively better excretion properties.
Finally, the selected compounds demonstrated similar or better overall pharmacokinetic parameters than that of curcumin, as observed with the ADME discussed above.
A combined pharmacophore model and molecular docking for virtual screening has been conducted to find a potential DYRK2 inhibitor.
Based on a gradual virtual screening process using a ligand-based pharmacophore model and molecular docking, 11 hit compounds have been selected. Further detailed study using molecular dynamics simulation afforded six hit compounds with better binding interaction with DYRK2 compared to that of curcumin, i.e. Molport-035-369-361 (-71.35±24.85) kJ/mol, Molport-000-004-273 (-83.56±18.24) kJ/mol, MolPort-029-697-986 (-61.69 ±14.89) kJ/mol, MolPort-035-585-822 (-74.24±15.55) kJ/mol, MolPort-000-156-336 (-54.49±15.09) kJ/mol, and MolPort-002-747-457 (-69.36±12.58) kJ/mol.
The six compounds obtained after a gradual virtual screening process have similar pharmacophore characteristics. Considering the pharmacokinetic properties, Molport-035-369-361, MolPort-035-585-822 as well as Molport-000-004-273 are now under in vitro study for further investigation as lead compounds, and the results will be reported elsewhere.
Protein Data Bank: DYRK2 data from Protein Data Bank. Accession number PBD5ZTN; https://identifiers.org/structure/5ztn.
Dataset of compounds with biological activity for ligand-based pharmacophore modeling were obtained from21 and are shown in Figure 1.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)