Keywords
Workflows, Containers, Cloud computing, Reproducibility, Automation, Big Data
This article is included in the Software and Hardware Engineering gateway.
This article is included in the EMBL-EBI collection.
Workflows, Containers, Cloud computing, Reproducibility, Automation, Big Data
API: Application programming interface
AWS: Amazon web services
CRD: Custom resource description
GUI: Graphical user interface
HPC: High-performance computing
NGS: Next-generation sequencing
PV: Persistent volume
PVC: Persistent volume claim
SFTP: SSH file transfer protocol
UI: User interface
VM: Virtual machine
VMI: Virtual machine image
WMS: Workflow management system
The life sciences have become data-intensive, driven largely by the massive increase in throughput and resolution of molecular data generating technologies. Massively parallel sequencing (also known as next-generation sequencing or NGS) is where the largest increase has been seen in recent years, but other domains are also increasing dramatically, including proteomics, metabolomics, systems biology and biological imaging.1-3 Consequently, the need for computational and storage resources has continued to grow, but the focus has also changed towards the downstream steps of biological experiments and the need to carry out efficient and reproducible data analysis. While high-performance computing (HPC) and high-throughput computing clusters remain the main e-infrastructure resource used in biological data analysis, cloud computing is emerging as an appealing alternative where, in the infrastructure-as-a-service case, scientists are able to spawn virtual instances or infrastructure on demand to facilitate their analysis, after which the resources are released.4,5 One area that has traditionally often caused headaches for users is the installation of software tools and their inclusion into workflow tools or other computing frameworks.6 This is an area where cloud resources provide an advantage over HPC, in that scientists are not dependent on system administrators to install software, but can handle the installation themselves. However, software for biological analyses can be quite challenging to install due to sometimes complex dependencies. Virtual machines (VMs) offer the benefit of instantiating ready-made environments with data and software, including all dependencies for specific tasks or analyses and constitute a big step towards making computational reproducibility easier and more realistic to achieve in daily work. VMs form the backbone of traditional cloud-based infrastructures. Such images can easily be shared between users and can be deposited and indexed by one of the available virtual machine image (VMI) catalogs.7 VMIs, however, can easily become large and take considerable time to instantiate, transfer or rebuild when software in the image needs to be updated.
Software container technology has emerged as a complement to VMs. While they offer a bit less isolation between processes, they are on the other hand more lightweight in terms of storage and memory requirements and are thus easier to share. By sharing the operating system kernel with the host computer, they are also much faster to launch and terminate.8 Containers encompass all dependencies of the provisioned tools and greatly simplify software installations for end users. The most widely used containerization solution is Docker (www.docker.com), but Singularity,9 uDocker10 and Shifter,11 are recent alternatives that prevent users from running containers with root privileges, thus addressing the most common security issue when deploying containers in multi-tenant computing clusters such as on HPC clusters. Docker containers are commonly shared via Docker Hub.12 There are also initiatives for standardizing containers in the life sciences such as BioContainers.13 Containers have seen an increased uptake in the life sciences, both for delivering software tools and for facilitating data analysis in various ways.14-18
When running more than just a few containers, an orchestration system is needed to coordinate and manage their execution and handle issues related to e.g. load balancing, health checks and scaling. Kubernetes19 has over the last couple of years become the de facto standard container orchestration system, but Docker Swarm20 and Apache Mesos21 are other well-known systems. A key objective for these systems is to allow the users to treat a cluster of compute nodes as a single deployment target and handle the packaging of containers on compute nodes behind the scenes.
While orchestration tools such as Kubernetes enable scientists to run many containers in a distributed environment (such as a virtual infrastructure on a cloud provider), the problem remains how to orchestrate the scheduling and dependencies between containers and being able to chain (define data dependencies between) them into an analysis pipeline. This is where scientific workflow management systems (WMSs) can be very useful; in fact, it can be difficult to carry out more complex analyses without such a system. Traditionally in data-intensive bioinformatics, an important task for WMSs has been to support analyses consisting of several components by running a set of command-line tools in a sequential fashion, commonly on a computer cluster. The main benefits of using a WMS include: i) making automation of multi-step computations easier to create, more robust and easy to change; ii) providing more transparency as to what the pipeline does through its more high-level workflow description and better reporting and visualization facilities; and iii) providing more reliable policies to handle transient or persistent error conditions, including strategies to recover from interrupted computations while re-using any partially finished data.
Using containerized components as the nodes in a scientific workflow has the advantage of adding isolation between processes and the complete encapsulation of tool dependencies within a container, and reduces the burden to administrate the host system where the workflow is scheduled to run. This means the system can be tested on a local computer and executed on remote servers and clusters without modification, using the exact same containers. Naturally, this opens up for execution on virtual infrastructures, even those provisioned on-demand with features such as auto-scaling. Apart from greatly simplifying access to the needed distributed compute resources, a positive side-effect is that the entire analysis becomes portable and reproducible.
Data ingestion and access to reference data constitute a key step when using cloud-based resources. When spawning a virtual infrastructure, data used in the analysis either needs to be available on storage connected to the compute nodes, or ingested into a locally provisioned file system. At the end of the analysis, the resulting data needs to either be put into persistent storage on the cloud or downloaded to local storage. However, the overhead with this can also be turned around. If data resides on storage connected to a cloud infrastructure, it is possible to simply move the workflow and containers there and “bring compute to the data”.
One key difference with running workflows locally on a desktop computer or server, or on a single HPC node, is that when running containerized workflows, the workflow engine often sits on another compute node than the executing containers. Also, the storage system is also typically separated, and accessible from any container, regardless of the exact compute node in a cluster where it is executed. Figure 1 illustrates this.
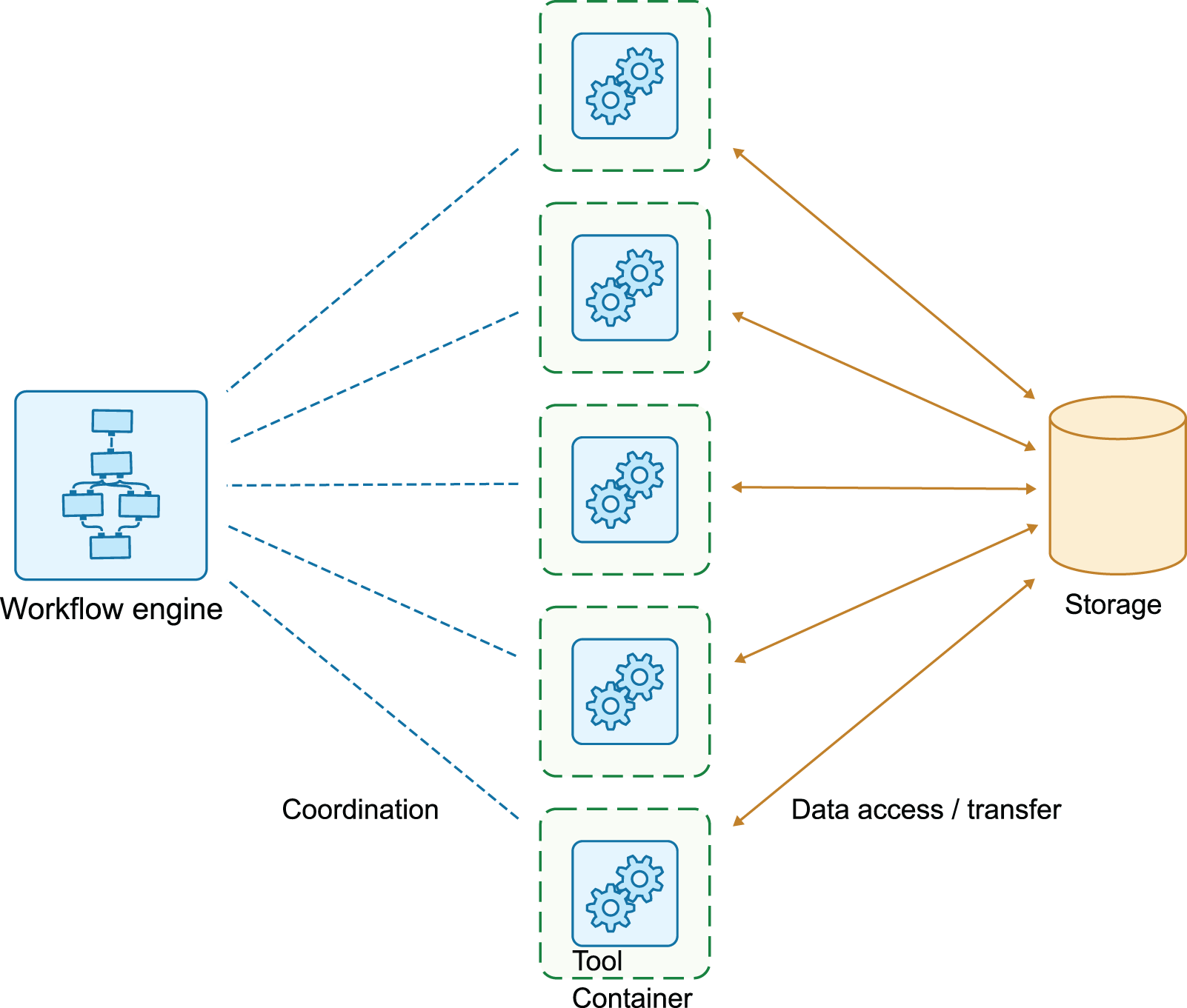
The tools pass data between each other by reading and writing to a shared storage system.
There are also multiple possibilities on how to run a workflow engine in relation to the containers, as the workflow engine can either be run on a separate system from the container cluster, such as a researcher’s local computer, or the workflow engine can itself run in a container on the cluster. Figure 2 illustrates this.
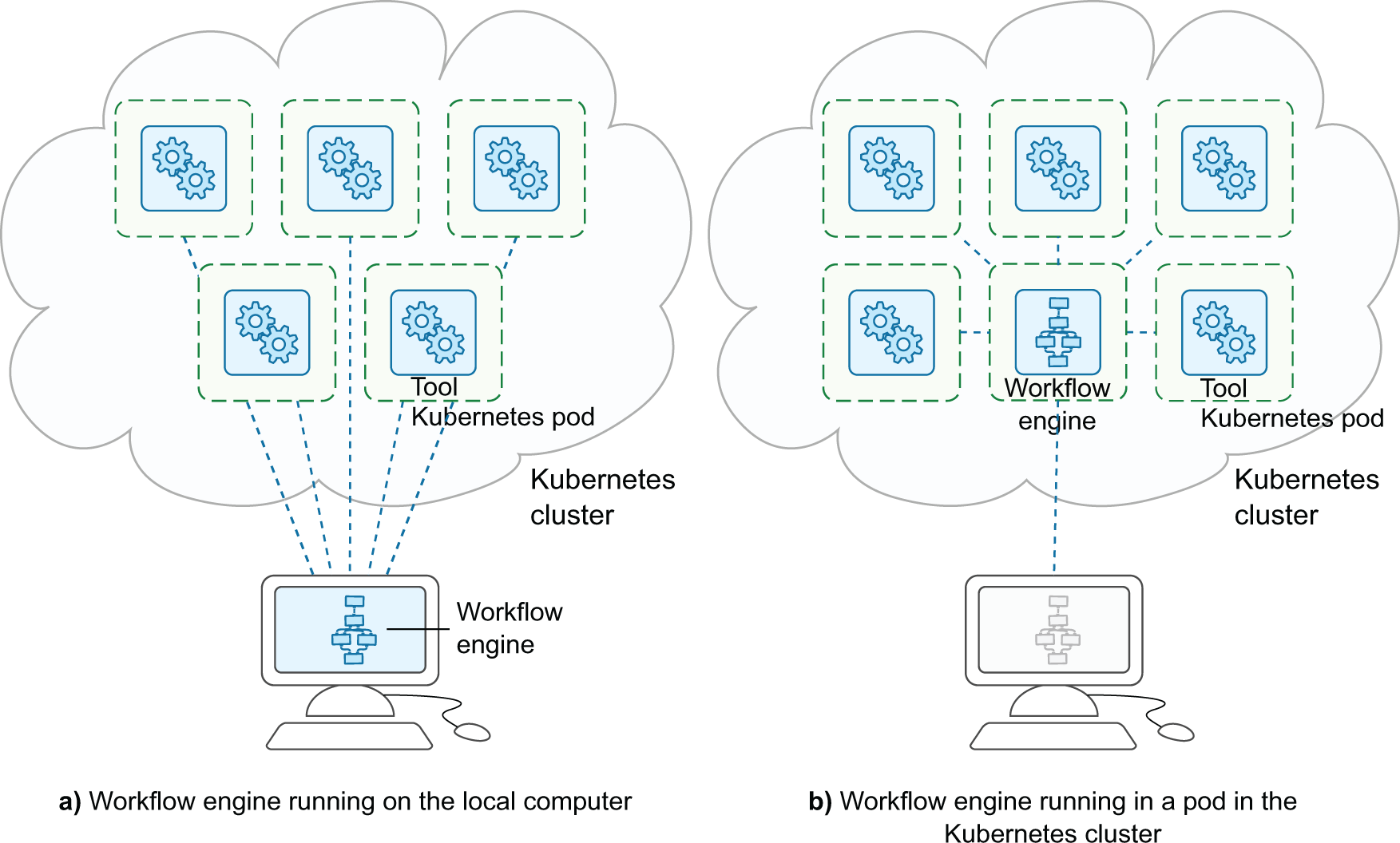
It can run either a) on the user’s computer, or b) in a pod in the Kubernetes cluster.
Due to their simplicity, containers are supported out of the box by most workflow tools. However, the approach for interacting with containers differs, especially when using virtual infrastructures on cloud resources. This section describes the approaches taken by a set of workflow management systems to interacting with containers in cloud environments, with varying degree of uptake in the life science community.
Nextflow22 is a workflow framework that aims to ease the implementation of scientific workflows in a portable and reproducible manner across heterogeneous computing platforms and to enable the smooth migration of these applications to the cloud. The framework is based on the dataflow paradigm, a functional/reactive programming model in which tasks are isolated from each other and are expected to be executed in a stateless manner. This paradigm simplifies the deployment of complex workflows in distributed computing environments and matches the immutable execution model of containers.
Nextflow provides built-in support for the most used container runtimes i.e. Docker, Singularity, Shifter and uDocker (though the last two are undocumented because of still being in an experimental state). The use of containers is defined in a declarative manner, i.e. by specifying the container image that a workflow needs to use for the task executions. Once annotated in this way, the Nextflow runtime takes care of transparently running each task in its own container instance while mounting the required input and output files as needed. This approach allows a user that needs to target the requirements of a specific execution platform to quickly switch from one containerization technology to another with just a few changes in the workflow configuration file (e.g. the same application can be deployed in an HPC cluster using Singularity or in the cloud with Docker).
Great flexibility is given for container image configuration. Nextflow supports both the image-per-workflow pattern (the same image for all executed tasks) and the image-per-task pattern (each task uses a different container image). The container image used by a task can even be specified at runtime by a given input configuration.
Nextflow allows the deployment of containerized workloads in a cloud-native manner using different strategies, as briefly discussed in the following paragraphs.
Cloud-managed computing service: in this approach workflow tasks are submitted to a cloud provider batch execution service. The containerization and auto-scaling are delegated to the cloud service. The workflow inputs/outputs and intermediate result files need to be stored using the provider object storage service. Nextflow adapts the task definition, resource requests and container image to the cloud provider format, submitting the corresponding API requests and properly staging the task data. Currently, Nextflow has built-in support for the Amazon Web Services (AWS) Batch service and the Google Genomics Pipelines service.
Cloud unmanaged computing service: when using this deployment strategy, Nextflow takes care of the provisioning of a set of cloud VM instances in which it automatically sets up its own clustering engine for the distributed execution of the workflow. The VM instances only require the availability of a Docker runtime and the Java virtual machine. Workflow tasks exchange data via a shared file system or by using a cloud object storage service. The workflow execution is managed by Nextflow which also handles the cluster auto-scaling, i.e. adds or removes VM instances on-demand to adapt the actual needs of a workload at any point in time and optimizes the overall computing cost when applicable. At the time of writing, this feature supports the use of the AWS EC2 cloud service and Google Compute Engine service.
Figure 3 provides an overview of the components involved when executing a containerized Nextflow workflow.
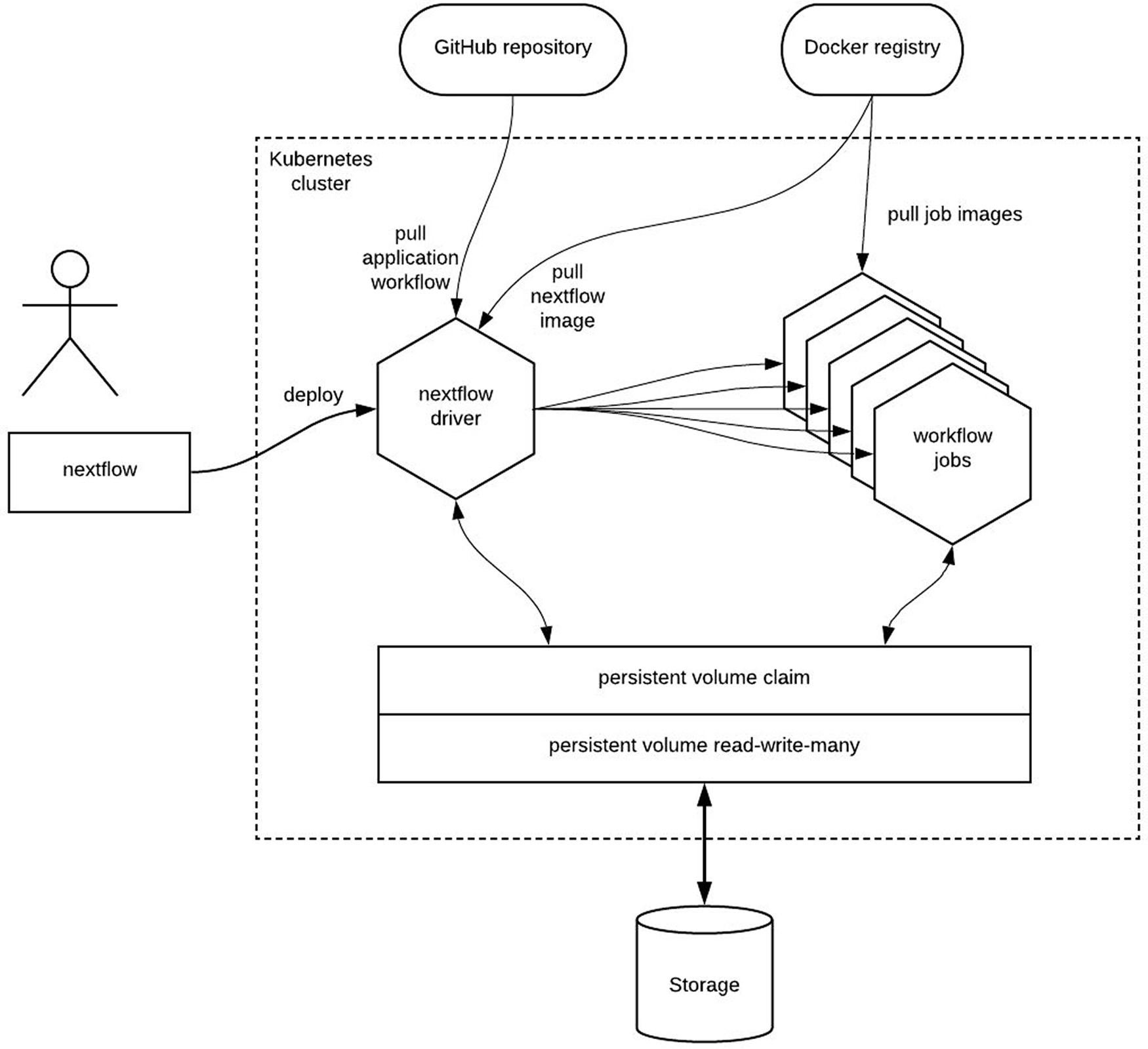
The tools pass data between each other by reading and writing to a shared storage system.
Galaxy (Galaxy, RRID:SCR_006281) is a data-analysis workflow platform that has probably the most active development community in the field of workflow environments in Bioinformatics. This has enabled it to persist as an active project and to continue to evolve since 2005, enabling reproducible research through active channels for sharing tools, workflows and datasets.23 Galaxy is currently used by tens of thousands of scientists across the globe.24 Besides offering a modern user interface (UI) for end users, Galaxy is accessible through its REST API or through the BioBlend Python binding25 (that talks to this REST API).
For execution versatility, Galaxy has adapters to offload jobs in a wide variety of systems, from local Docker containers to different batch scheduler systems. In Galaxy, there is a complete separation of concerns between tools, workflows and execution environments, meaning that a workflow definition is not bound to a particular execution environment. Within the last seven years Galaxy gained the ability to launch on AWS through the CloudMan Launcher Galaxy sub-project.25 This initial setup followed a relative VM centric approach, which has natural limitations for scaling in the number of tools included as they all need to be provisioned inside the VM used for the provisioned cluster.
More recently, through efforts within the PhenoMeNal H2020 Project,26 and later contributions by the Genomics Virtual Lab (GVL) initiative, Galaxy gained the ability to deploy inside Kubernetes in an automated fashion through the use of Helm Charts.27 Since then, the Galaxy Helm chart has been adopted as a project by the Galaxy community (https://github.com/galaxyproject/galaxy-helm), and two major versions have been released, making v3.x of the Galaxy Helm Chart a mature product relying on a minimal Galaxy container for fast deployment. Galaxy Helm Chart v3 allows the deployment of multiple web and jobs handlers, making the setup scalable from any perspective (number of user and number of jobs). Newer versions of the CloudMan Galaxy launcher now rely on this setup instead of VM deployments and recently a Single Cell RNA-Seq Galaxy setup28 was released based on the Galaxy Helm chart.
The Kubernetes job runner, also contributed to the Galaxy community by the PhenoMeNal H2020 Project and then further maintained by the Galaxy community, allows Galaxy to offload jobs to Kubernetes, either when Galaxy runs inside the container orchestrator or from the outside (provided that the shared file system is accessible to both the container orchestrator and Galaxy). The Kubernetes runner for Galaxy will take care of resource requirement settings, recovery of jobs in Kubernetes if there is a restart, auto-upgrading of resource utilization, Persistent Volume Claim mounting both for Galaxy and in each jobs pod, as well as adequate supplementary groups handling for network filesystem and other shared file systems. This integration requires the use of a shared file system, which needs to be mountable in Kubernetes in a read-write-many configuration, as both the main Galaxy job handler pod and the job pods need to mount it at the same time (through the PV/PVC abstraction). The Kubernetes runner for Galaxy adheres to the principles of resilience and redundancy commonly considered in cloud computing, providing ways to recover from jobs being lost by cluster nodes going down in the middle of an execution.
The Kubernetes runner for Galaxy can benefit from explicit tool-to-containers mapping through Galaxy dynamic destinations, or take advantage of dynamic bioconda-to-containers mapping thanks to built-in functionality in Galaxy for this purpose. This means that if a Galaxy tool declares a Bioconda package as the dependency to be resolved, it will bring an automatically built container for that Bioconda package29 from BioContainers.13 For tools with multiple conda packages as dependencies, the Galaxy/Biocontainers community builds automatically multi-tool “mulled” containers (https://github.com/BioContainers/multi-package-containers) through Planemo-Monitor (https://github.com/galaxyproject/planemo), and these are as well resolved automatically when running on the Kubernetes setup.
The Galaxy-Kubernetes integration, which includes all the aspects related to Kubernetes mentioned in the previous paragraphs, has been battle tested by the PhenoMeNal H2020 Project, having its public instance receiving in the order of tens of thousands of jobs during the past 4 years, having tens of deployments in production environments and possibly hundreds of deployments in development settings. It has also been used as part of GVL’s CloudMan for the past year and for running training courses with more than 30 concurrent users in the context of Single Cell RNA-Seq. This integration has been tested locally on Minikube, on OpenStack deployments, on Google Cloud Platform and AWS turn-key Kubernetes services. In these two last cases, the setup can behave elastically, deploying more Kubernetes worker nodes automatically for increased capacity on demand, as the number of jobs or users increment, and then automatically reducing the number of nodes and containers deployed when load diminishes. It is expected that in the next few years the Galaxy-Kubernetes integration is embraced in the areas of proteomics and transcriptomics, and becomes one of the main routes of deploying Galaxy installations.
Pachyderm30 is a large-scale data processing tool built natively on top of Kubernetes. This platform offers: i) a data management system based on Git semantics and ii) a workflow system for creating distributed and reproducible pipelines based on application containers. In order to create workflows with Pachyderm, users supply a JSON pipeline specification including a Docker image, an entrypoint command to execute in the user containers and one or more data input(s). Afterward, Pachyderm will ensure that the corresponding pods are created in Kubernetes, and will share the input data across them and collect the corresponding outputs. Thanks to using Kubernetes for container orchestration, Pachyderm is able to, among other things: i) optimize cluster resource utilization, ii) run seamlessly on multiple cloud and on-premise environments, iii) self-heal pipeline jobs and related resources.
This workflow tool provides an easy mechanism to distribute computations over a collection of containers. In other words, it offers similar capabilities to frameworks such as Apache Spark, but replaces MapReduce syntax with legacy code. More specifically, Pachyderm is able to distribute workloads over collections of containers by partitioning the data into minimal data units of computation called “datums”. The contents of these datums are defined by glob patterns such as “/” or “/*”, which instruct Pachyderm how to process the input data: all together as a single datum, or in isolation (as separate datums). Users can define the number of pipeline workers to process these datums, which will be processed one at a time in isolation. When the datums have been successfully processed the Pachyderm daemon gathers the results corresponding to each datum, combines them and versions the complete output of the pipeline. The scheduling of pipeline workers is determined by the resource utilization status of the workers’ nodes and the resource requests that are made for each pipeline, allowing for efficiently distributing workloads. Recently, work to integrate and demonstrate Pachyderm in bioinformatics was published.31
Luigi32 is an open-source Python module for defining and running batch-like workflows. The system is strongly oriented towards Big Data, and it comes as a convenience tool to pipe jobs from well-established analytics frameworks (e.g. Hadoop, Spark, Pig and Hive). Hence, instead of trying to substitute any of the available analytics systems, Luigi provides the means to stitch together jobs from different frameworks in a single batch-oriented workflow. In doing so, Luigi seamlessly handles many boilerplate operations such as: dependency resolution, visualization, failure tolerance and atomic file system operations.
The Kubernetes integration was developed by the PhenoMeNal consortium, and it is now part of the official Luigi project and thus being supported and maintained by the community. The integration enables to include pipeline processing steps as Kubernetes Jobs, which represent batch-like application containers that run until command completion. Hence, Luigi mainly uses Kubernetes as a cluster-resource manager where multiple parallel jobs can be run concurrently. To this extent, it is important to point out that Luigi is not meant to be used as a substitute for parallel processing frameworks (such as Spark and Hadoop), as it has limited support for concurrent tasks. In fact, in our experience Luigi can handle up to 300 parallel tasks before it starts to break down, or 64 parallel tasks if running on a single node. A project to adapt Luigi for scientific applications in general and bioinformatics in particular resulted in a tool named SciLuigi.33
We benchmarked Luigi on Kubernetes by reproducing a large-scale metabolomics study.34 The analysis was carried out on a cloud-based Kubernetes cluster, provisioned via KubeNow and hosted by EMBL-EBI, showing good scalability up to 40 concurrent jobs.35 Workflow definition, and instructions to reproduce the experiment, are publicly available on GitHub.36
SciPipe37 (SciPipe, RRID:SCR_017086) is a workflow library based on Flow-based programming principles, which enables building workflows from component libraries of predefined workflow components. SciPipe is implemented as a programming library in Go, which enables using the full power of the Go programming language to define workflows, as well as to compile workflows to executable files, for maximum ease of deployment in cloud scenarios.
SciPipe has been used to orchestrate machine learning pipelines to build predictive models for off-target binding in pharmacology.38 SciPipe currently provides experimental support for container based workloads via Kubernetes, implemented through the official Kubernetes Go client library.39 The Go client library enables transparent access to a Kubernetes cluster regardless of whether the SciPipe workflow runs outside the cluster or inside, in a pod. When running inside the cluster, it automatically detects cluster settings. When not inside a cluster, the connection to a cluster can be configured by setting the appropriate environment variables. The Kubernetes integration in SciPipe is demonstrated through the implementation of a use case workflow consisting of a set of data preparation steps for mass-spectrometry data, using the OpenMS software suite.40 It contains a Go file (with the .go extension) with the workflow, and a Kubernetes job-spec file in YAML format (with the .yml extension) for starting SciPipe inside a Kubernetes cluster is together with the workflow.41
Singularity containers9 can already today be used from SciPipe on a simple level, by configuring the appropriate shell commands when creating new processes through the shell-command process constructor (the scipipe.NewProc() function in SciPipe). Development of more integrated support for Singularity containers is planned.
Argo workflow42 originates from a corporate application with the need of a competent continuous integration/continuous deployment solution but matured into a generalized workflow engine. It was open sourced by Intuit and is now adopted across several industries and companies among them life science and biopharma. A big differentiator of Argo is that no domain specific language knowledge is required to build workflows. The workflows are instead constructed by a clearly defined declarative approach manifested as Kubernetes custom resource definitions (CRDs), which is a templated approach to enable composability and extendability to the original Kubernetes resource orchestration. Argo is often referred to as a cloud native workflow orchestration tool and is designed for container native execution without the overhead of other non-native container solutions. Argo provides a robust way to declare and run workflows as arguments and artifact objects can be declared as input and output and containers run as state transitions merely passing objects and arguments through the pipeline. In each workflow step the state is stored in redundant etcd43 memory for resilience and reproducibility. Argo is complemented by an artifacts concept that enables data and parameter versioning through complementing Argo with an S3 compatible storage backend. Furthermore Argo events enable signals and events for triggering workflows on webhooks, resource changes, GitHub or Docker image changes etc enabling additional automation scenarios.
Kubeflow44 is another open-source platform for Kubernetes, specifically aimed at building and running machine learning workflows and is developed by Google. As a framework, Kubeflow is intended to provide a set of tools for various tasks in the process of machine learning workflows, from data preparation and training to model serving and maintenance. Many of the tools are existing tools with a user base in the industry, such as Jupyter Notebook and Seldon, while others are customized wrappers or endpoints to frameworks like TensorFlow, PyTorch.
In particular, Kubeflow provides a workflow tool called Kubeflow Pipelines, which allows users to create workflows of varying complexity, chaining together series of container operations. The tool is based on the workflow engine Argo, which has been integrated with the Kubeflow ecosystem with a different UI than the original Argo UI. In essence, a user declares a workflow in the Kubeflow Pipelines software development kit, defining the different steps in terms of container operations, i.e. what container to use for the step, Kubernetes resource allocations, commands to execute in the container and so on. The user also declares the order of these container operations and any desired input parameters to the workflow, and from this definition a pipeline can be compiled. This pipeline is a portable configuration which in theory can be uploaded and used in any Kubeflow environment. With the pipeline deployed in Kubeflow, a user can start a run where available pipeline parameters can be provided and the progress of the run tracked in the Kubeflow Pipelines UI. Here, logs of the container’s standard output, output artifacts and various Kubernetes information can be tracked for each step, along with a visualization of the workflow graph.
As an evaluation of the Kubeflow Pipelines system, a simple pipeline was developed, based on previous work45 where a convolutional neural network is trained to “… predict cell mechanisms of action in response to chemical perturbations...”, based on a dataset of cell microscopy images from the Broad Bioimage Benchmark Collection. The pipeline covers the following steps of the machine learning process: data pre-processing, model training, model evaluation and model serving preparation. The result is a workflow that handles the entire process end-to-end, building a servable machine learning model and publishing this as a Docker container that can be deployed for serving predictions. The pipeline source code, while partly tailored to a specific environment, is available in.46
The tools covered in this study have taken slightly different approaches to work with containers, with different implications. Most tools are designed to work with general command-line tools while also supporting containers, except Pachyderm, Argo and Kubeflow that are Kubernetes-native and only support containers as means of processing.
Workflow tools can almost always work with containers instead of command-line tools, using the docker run command, and taking advantage of an external scheduling system such as a batch scheduler (e.g. SLURM). When deployed in cloud environments, Kubernetes is the most widely used orchestration framework by the tools, though Galaxy also supports Docker Swarm as well and provides a convenient mapping from the Conda package manager to containers. For Kubernetes, tools differ in the way they interact with its API; Pachyderm, Argo, Kubeflow Pipelines and SciPipe interact via the Go API whereas Nextflow, Galaxy and Luigi communicate via the REST API. While these APIs are not identical, the API designs are very similar in terms of data structures and in effect allow for the same level of control. In more detail, the structure of the JSON documents sent to the REST API is closely matched by the hierarchic structure of the ‘spec’ structs in the Go API. The workflow tools also differ somewhat in the level of flexibility they allow for utilizing the API features, where Luigi is the most versatile as it allows for passing a raw JSON string with full flexibility but also requires the most skills from workflow authors.
Error management for containerized workflows is an important concept. Kubernetes is designed to restart pods that fail, which is not particularly useful if there is an error reported from the containerized tool. The cloud paradigm includes fault-tolerance, and the tools differ in their strategies. Galaxy, Luigi and SciPipe use Kubernetes Jobs, which allows restarting the pods N times before reporting as failure. Argo and Kubeflow Pipelines run workflow steps as pods that are governed by Argo’s custom Kubernetes resource workflow; as such errors for individual containers are handled as for normal Kubernetes pods, including error messages detailing reasons for failure etc. Nextflow offers the possibility to automatically tune the job, e.g. to increase the available memory for a job before reporting it as failed.
The way in which workflow systems schedule containers on Kubernetes is one factor where they differ from each other. Nextflow implements an actor-like model to handle job requests. Jobs are submitted in a manner similar to an internal job queue. Then a separate thread submits a request to the Kubernetes cluster for each of them and periodically checks their status until the job completion is detected. Galaxy can deploy an unlimited number of job handlers, that can each iterate over thousands of jobs, and are separate processes to the web server handlers. It keeps no separate threads or processes per Kubernetes job. Pachyderm makes direct use of Kubernetes scheduling and uses etcd for storing metadata for data versioning and status of all jobs. It is also the only system that uses the Kubernetes feature of “parallel jobs”. Argo and Kubeflow Pipelines similarly utilize etcd for storing metadata of workflows and executions, and employ a workflow controller component to schedule containers based on the state of deployed workflows. Luigi keeps a thread open and continuously polls the job status. SciPipe keeps a lightweight thread (go-routine) alive for each created job, as the Go API call for creating jobs do block the calling go-routine until the job finishes or is stopped for other reasons.
Handling many concurrent jobs can become a problem in Luigi/SciLuigi, where a separate Python process is started for each job which imposes a practical limit on the number of concurrent jobs; in the authors’ experience 64 is a rule of thumb for an upper limit if running on a single node, or else 300. Going above this tends to result in HTTP timeouts since workers are talking to the central scheduler via HTTP calls over the network.
So what are the main differences between the systems when running containers in cloud environments? Galaxy has a graphical user interface and might be more attractive for users with less expertise in programming/scripting, although it still provides programmatic access (REST and Python bindings) for workflow/tool execution and data/metadata retrieval. It can be run on both HPC batch schedulers and on cloud. Workflows in Argo can be submitted and controlled both from multi platform command-line interfaces as well as monitored through a graphical UI (GUI) but can also be controlled by other Kubernetes cluster resources since workflows are standard CRD resources. Kubeflow also provides a GUI as the main method for running pipelines, in addition to being a very portable platform in Kubernetes environments. Kubeflow Pipelines does however have a somewhat steep learning curve as the Python-based domain-specific language requires a good grasp of Kubernetes concepts. Nextflow, just like Galaxy (and Snakemake47), provides some versatility when working with containers, as being able to operate both on cloud or HPC batch schedulers. Pachyderm, Argo and Kubeflow are built specifically for running on Kubernetes and Pachyderm has a data-versioning file system built-in. Argo and Kubeflow Pipelines support artifacts in workflows and versioning of input and output artifacts through interfacing with an optional S3 storage backend. Luigi/SciLuigi supports many data sources out of the box, including HDFS, S3, etc and integrates seamlessly with Apache Hadoop and Spark. SciPipe implements an agile workflow programming API that smoothly integrates with the fast growing Go programming language ecosystem.
Finally, perhaps somewhat surprisingly, we would like to end with a word of caution regarding the use of containers. While containers have many advantages from a technical perspective and alleviate many of the problems with dependency management, it also comes with certain implications, as it can make tools more opaque and discourage accessing and inspecting the inner workings of the code inside the container. This could potentially lead to some lack of understanding of what a specific tool does. Thus, when wiring together containers in a scientific workflow, proper care needs to be taken that the observed output matches what would be expected from using the tool directly.48 Also for this reason we strongly suggest to follow community best practices for packaging and containerization of bioinformatics software.49
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)