Keywords
Mycobacterium tuberculosis, Serine/Threonine Protein Kinases A, B, G, Molecular Docking, Molecular Quantum Similarity Analysis, Electrostatic Interactions.
This article is included in the Cheminformatics gateway.
Mycobacterium tuberculosis, Serine/Threonine Protein Kinases A, B, G, Molecular Docking, Molecular Quantum Similarity Analysis, Electrostatic Interactions.
Protein kinases (PKs) are a key in controlling proliferation and differentiation in eukaryotic cells in living organisms. They are enzymes that catalyze the protein phosphorylation process. An important reason to analyse the protein phosphorylation process is that it represents an attractive drug target in a variety of fatal diseases such as cancer1. The PKs present in the human body have been widely used in the field of drug design because they play an important role as therapeutic targets in these diseases. However, little is known about the serine/threonine protein kinases (STPKs) involved in tuberculosis. For this reason, we propose a study about crystalized STPKs Pkn A, B and G, to gain new insights in the mechanism of the phosphorylation process.
To date, one of the main questions in the tuberculosis field is ‘what are the protein substrates of the STPKs?’ According to Av-Gay et al.2, to deal with this topic we need to study the interactions of crystalized STPKs Pkn A, B and G with a particular series of ligands to each PK. The competitive ATP ligands used are a series of compounds for Pkn A reported by Sipos et al.3, ligands for Pkn B reported by Székely et al.4, Loughheed et al.5 and Chapman et al.6 and ligands for Pkn G reported by Sipos et al.3 and Pato et al.7. These ligands were selected with the goal of finding new insights into the inhibition of oxidative phosphorylation. Therefore, we are researching new pharmacological alternatives for this disease through the structural models of the ligands on the ATP sites.
Another aim of this study is to conduct molecular quantum similarity (MQS) analysis using a quantum similarity field to understand the electronic and structural patterns involved in stabilization of the active site of the inhibitors for each PK studied. Therefore, a hybrid quantum mechanics/molecular mechanics (QM/MM) approach was used to carry out a structure-activity relationship analysis of the inhibitor set for Pkn A, Pkn B and Pkn G.
The crystal structures of Pkn A (PBD code: 4OW8, resolution: 2.03 Å8), Pkn B1 (PDB code: 3F69, resolution: 2.80 Å9(a)), Pkn B2 (PDB code: 1MRU, resolution: 3.0 Å9(b)), Pkn B3 (PDB code: 2FUM, resolution: 2.89 Å9(c)) and Pkn G (PBD code: 2PZI, resolution: 2.40 Å10) were selected for the preparation process using Schrödinger Suite 2014-1’s Protein Preparation Wizard module11, which refines the protein structure and optimizes the hydrogen bond (H-bond) network. The open-source software AutoDock Vina could also be used to perform these steps.
Protonation states were identified using PropKa utility at a physiological pH12. After, restrained molecular minimization using the Impact Refinement (Impref) module13 was carried out, taking into account the heavy atoms restrained within a root-mean-square deviation (RMSD) of 0.18 Å from the initial coordinates.
The main characteristics of Pkn A and B consist of a transmembrane receptor with a tyrosine kinase domain protruding into the cytoplasm. For Pkn G, the unique multidomain topology of this PK reveals a central kinase domain that is flanked by N- and C-terminal rubredoxin and tetratrico-peptide repeat domains, respectively13,14.
The molecular datasets used for the docking study were reported by Sipos et al.4, Székely et al.5, Lougheed et al.6, Chapman et al.7 and finally Pato et al.8 (see Table 1–Table 6). To select this molecular dataset, the structural diversity and uniform distribution of IC50 was taken into account. Logarithmic IC50 (μM) (pIC50=−log IC50) was employed as the dependent variable instead of IC50. The 3D molecular structures of the compounds were drawn and optimized using Maestro15 and then prepared with the LigPrep module16, where ionization/tautomeric states were predicted at physiological pH conditions using Epik17. Afterwards, energy minimization was performed using Macromodel18 with the OPLS2005 force field. The open-source software AutoDock Vina could also be used to perform these steps.
| Compounda | Structure | pIC50 |
|---|---|---|
| 1 |  | -1.568 |
| 2 |  | -1.839 |
| 3 |  | -1.875 |
| 4 |  | -1.934 |
aCompounds to Pkn A reported by Sipos et al.4
| Compound | Structure | pIC50 |
|---|---|---|
| 5a,b |  | -0.585 Pkn G:1.301 |
| 6a |  | 0.569 |
| 7a |  | 0.086 |
| 8a |  | 0.585 |
| 9a |  | 0.237 |
| 10c |  | -0.375 |
| 11d |  | 1.276 |
| 12d |  | 1.252 |
| 13d | 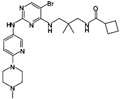 | 1.046 |
| 14d |  | 0.285 |
| 15d |  | -1.219 |
| 16d |  | -0.1758 |
| 17d |  | 0.462 |
| 18d |  | 1.259 |
| 19d |  | 1.017 |
| 20d |  | 1.036 |
| 21d |  | -0.792 |
| 22d |  | 1.187 |
 |  | ||
| Compounda | R1 | NHR2 | pIC50 |
|---|---|---|---|
| 23 |  |  | 0.492 |
| 24 |  |  | 0.344 |
| 25 |  |  | -0.065 |
| 26 |  |  | 0.652 |
| 27 |  |  | 0.357 |
| 28 |  |  | 0.971 |
| 29 |  |  | 0,369 |
| 30 |  | - | 0.426 |
| 31 |  | - | 0,416 |
| 32 |  | - | 0.406 |
| 33 |  | - | 1.066 |
| 34 |  | - | 0.618 |
| 35 |  | - | 1.060 |
| 36 |  | - | 0.695 |
| 37 |  | - | 1.076 |
| 38 |  | - | 0.451 |
| 39 |  | - | 0.146 |
| 40 |  | - | 1.130 |
| 41 |  | - | 1.194 |
| 42 |  | - | 0.664 |
| 43 | 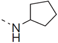 | - | 0.939 |
aCompounds to Pkn B reported by Chapman et al.7
 |  | ||
| Compounda | NR1R2 or OR1 | R3 | pIC50 |
|---|---|---|---|
| 44 |  | - | 1.056 |
| 45 |  | - | 0.917 |
| 46 |  | - | 1.260 |
| 47 | 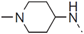 | - | 1.137 |
| 48 |  | - | 1.276 |
| 49 |  | - | 1.252 |
| 50 |  | CN | 1.252 |
| 51 | 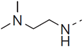 | CN | 1.409 |
| 52 |  | CN | 1.638 |
| 53 |  | SO2Me | 1.398 |
aCompounds to Pkn B reported by Chapman et al.7
 | |||
| Compounda | R1 | R2 | pIC50 |
|---|---|---|---|
| 54 | H | CN | -0.299 |
| 55 | OMe | H | -0.053 |
| 56 | Cl | H | 0.754 |
| 57 | CF3 | CN | 1.398 |
aCompounds to Pkn B reported by Chapman et al.7
 |  | ||||||||
| C | R1 | R2 | R3 | R4 | R5 | X | Bond C3-X | Bond C1-C2 | pIC50 |
|---|---|---|---|---|---|---|---|---|---|
| 58a | cyclopropyl | H | H | H | NH | C | C3-X | C1-C2 | 0.523 |
| 59a | cyclohexyl | H | - | H | NH | O | C3-X | C1-C2 | 0.167 |
| 60a | cyclopropyl | H | - | H | NH | O | C3-X | C1-C2 | 0.276 |
| 61a | isopropyl | H | -OH | H | NH | C | C3=X | C1=C2 | 1.523 |
| 62a | cyclopropyl | Br | -OH | H | NH | C | C3=X | C1=C2 | 1.699 |
| 63a | methy-cyclopropyl | Cl | -OH | H | NH | C | C3=X | C1=C2 | 1.699 |
| 64a | methy-cyclopropyl | Cl | -OH | H | NH | C | C3=X | C1=C2 | 1.699 |
| 65a | cyclopropyl | Cl | -OCH3 | Cl | NH | C | C3=X | C1=C2 | 1.398 |
| 66a | 1,3-benzodioxole | Cl | -OH | H | NH | C | C3=X | C1=C2 | 2.000 |
| 67a | methy-cyclopropyl | H | -OH | H | NH | C | C3=X | C1=C2 | 1.301 |
| 68a | methy-cyclopropyl | H | -OH | H | NH | C | C3=X | C1=C2 | 1.301 |
| 69b | p-methylbenzoate | H | H | H | NH | C | C3-X | C1-C2 | 0.699 |
| 70b | isopropyl | H | H | H | NH | C | C3-X | C1-C2 | -0.478 |
| 71b | Cyclobutyl | H | H | H | NH | C | C3-X | C1-C2 | -0.477 |
| 72b | methyl-isopropyl | H | H | H | NH | C | C3-X | C1-C2 | -1.176 |
| 73b | cyclopropyl | H | -Me | H | NH | C | C3-X | C1-C2 | 0.046 |
| 74b | -Me | H | -Me | H | NH | C | C3-X | C1-C2 | -1.079 |
| 75b | cyclopropyl | H | H | H | NH | S | C3-X | C1-C2 | -0.301 |
| 76b | Furanyl | H | H | H | C2H4OH | C | C3-X | C1-C2 | -1.204 |
| 77b | cyclopropyl | H | H | H | OH | C | C3-X | C1-C2 | -1.279 |
Docking studies were performed with the software Glide18 using standard precision (SP) mode and default parameters. The open-source software AutoDock Vina could also be used to obtain similar results. The docking grid was generated with standard protocol centered at the co-crystallized ligand. Due to that, the scaling factor for the van der Waals interactions is very important it was taken as 0.8. The radii of nonpolar protein atoms were selected to allow the binding of larger ligands. Further, the induced fit docking (IFD) workflow19 was also used to generate a good number of conformations of the receptor suitable for binding ligands that had strange orientations. It allows the protein to undergo side-chain movements, backbone movements, or both, upon ligand docking. The docking procedure has four steps where accuracy is ensured by Glide’s scoring function and Prime’s:
(i) The first step for Glide docking is carry out into the rigid receptor to generate a good ensemble of poses.
(ii) Analysis of the protein using Prime21. Taking into account side-chain prediction module followed by a structure energy minimization of each protein/ligand complex pose.
(iii) Analysis through re-docking of the ligand into low energy induced-fit structures taking in account the previous step using default Glide settings (no vdW scaling).
(iv) Finally, the binding energy is estimated using (IFDScore) by accounting for the docking energy (GScore), as well as receptor strain and some solvation terms (Prime energy).
To obtain the docking outcomes, the dynamics simulation of 100ns in vacuo using the GROMOS11 force field set was used on the protein–ligand complex, using Gromacs 5.1.220 to analyze the stability of the protein–ligand complexes22.
In recent studies, MQS methodology has shown good results in understanding steric and electronic effects of ligands23,24. The MQS field was introduced by Carbó and co-workers 30 years ago25–30 and has been used to understand the steric and electronic effect on the molecular sets. The mathematic relationship to obtain quantum similarity ZAB between two compounds A and B, with electron density ρA(r1) and ρB(r2) is defined using the minimization of the expression for the Euclidean distance as:
Therefore, the Euclidean distance is the square root of the integral on the squared differences between the densities ρA(r1) and ρB(r2). In this context, ZAB represents the similarity measure between the electronic densities of the compounds A and B, and ZAA and ZBB are the self-similarity of compounds A and B31. The similarity index very often used is the Carbó index31. This index is expressed as follows:
In Equation 2, (Ω) represents an operator. A simple way to make quantum similarity measures (QSM)29–31 involving two density functions, in the most usual form, is expressed as an integral:
In this equation ρA(r1) and ρB(r2) are the density functions to quantum objects A and B, while Ω(r1,r2) is a positive operator that defines weight. The Dirac delta function: δ(r1 – r2) is used to obtain the overlap similarity measure and to measure the Coulomb similarity the Coulomb operator: |r1 – r2|-1 is used. These operators are the most useful for obtaining similarity comparisons between ligands.
An important factor when carrying out the QSM is an optimal molecular alignment. As the integrals associated to the QSM produce, in any case, real positive results, the relative position problem can be approached using a maximal QSM. An overlap QSM can be expressed according to the equation:
In this equation, it is implicitly supposed that ρB(r) is translated and rotated by the six possible ways: (T;φ) and are shown as explicit parameters in this integral31. This key factor was applied using the topo geometrical superposition algorithm (TGSA)31.
The calculations of this study were performed with the GAUSSIAN 09 program32. The open-source software AutoDock Vina could also be used to obtain similar results. The structures were optimized using the B3LYP functional (DFT exchange-correlation method)33,34 and the 6-31G(d,p) basis set35. The contour lines of the molecular electrostatic potential have been obtained with the GaussView program36.
The conformational analysis of the STPKs defines the active and inactive form. These forms are also known as the DFG-in and DFG-out states. They are distinguishable by a flip of the Asp and Phe orientation in the activation loop as well as conformational changes in the surrounding loops37. The main difference between DFG-in and DFG-out conformations is that the different position of the DFG residue in the “out” form results in the opening of an additional cavity, the allosteric site, which can be hydrophobic in nature according to Zuccotto et al.38. In Pkn A the Asp side chain is buried and the Phe side chain is flipped outside the pocket. Therefore, Pkn A becomes DFG-out. However, in the STPKs Pkn B1 (crystal: 1MRU), Pkn B2 (crystal: 2FUM) and Pkn B3 (crystal: 3F69), the Asp side chain is exposed and the Phe side chain is buried (DFG-in), allowing ATP binding. Pkn G has DLG (Asp-Leu-Gly) instead of DFG (see Figure 1); the conformation is DLG-in. An important residue of STPKs is the gatekeeper. Their size determines the access of small, medium and large inhibitors to the back cavity even from the DFG-in state. In these crystallized PKs, the gatekeeper residues are MET95 for Pkn A, MET92 for Pkn B1 (crystal: 3F69), Pkn B2 (crystal: 1MRU), Pkn B3 (crystal: 2FUM), and MET232 for Pkn G. The methionine is a medium gatekeeper residue, according to Zuccotto et al.38 and allows access to small, medium and large inhibitors. This characteristic is important because it increases the range of molecular diversity in the development of new inhibitors.

DFG conformational analysis and gatekeeper residues to STPKs crystalized from Mycobacterium tuberculosis for (A) Pkn A, (B) Pkn B and (C) Pkn G. Hydrogen-bond-acceptor map (red mesh) for (D) Pkn A, (E) Pkn B and (F) Pkn G. Hydrogen-bond donor map (blue mesh) for (G) Pkn A, (H) Pkn B and (I) Pkn G. Hydrophobic map for (J) Pkn, (K) Pkn B and (L) Pkn G. Note: map sites were calculated using SiteMAP in Maestro. The open-source software AutoDock Vina could also be used to obtain similar results.
In Figure 1D, 1E and 1F we can see the hydrogen-bond-acceptor maps in red mesh. Pkn A shows a small red contour on the hinge zone (Figure 1D), a medium contour near to the DFG motif and a large contour near to the helix-αC and C-terminal loop. This is unlike Pkn B, which, using the crystal 3F69 (Figure 1E), shows only acceptor maps near to the DFG motif and in the C-terminal loop. Pkn G (Figure 1F) has large acceptor contours involving the hinge zone, DLG zone, helix-αC and C-terminal loop. Figure 1G, 1H and 1I shows the hydrogen-bond donor maps in blue mesh. In Pkn A (Figure 1G) we can see medium contours to hydrogen-bond donor on the hinge zone and C-terminal loop and a large contour near the DFG motif. In contrast, Pkn B (Figure 1H) shows only a large contour near to the DFG motif. Pkn G (Figure 1I) has small contours to hydrogen-bond donors along the hinge zone and near the DLG motif, helix-αC, C-terminal and N-terminal loops.
Analyses of hydrophobicity are shown in Figure 1J, 1K and 1L. Figure 1J shows a large contour of hydrophobicity for Pkn A near to the hinge zone and small contours on the C-terminal loop. Pkn B (Figure 1K) has only a small contour on the C-terminal loop. In contrast, Pkn G (Figure 1J) shows a large contour near the hinge zone, DLG motif and small contours near to the helix-αC and C-terminal loop. These physical-chemistry properties are important to describe the non-covalent stabilization on the active sites and selectivity of the ligands in the active pocket.
To obtain the docking outcomes, hydrogen bonds on the hinge zone and non-covalent interactions near the “gatekeeper door”, helix-αC, C-terminal and N-terminal were analyzed. An important insight are the non-covalent interactions. Theses interactions involved backbone, side chain hydrogen bonds and aromatic-aromatic interactions. On the other hand, the ligands with high scores have these characteristics in their non-covalent interactions. The ligands with lower scores have few to no interaction forces. Further, some of the higher scoring ligands forming hydrogen bonds and aromatic-aromatic interactions with the amino acid residues are related to the hinge zone.
Figure 2 shows the docking interactions of the Pkn A inhibitors from Table 1. These compounds have groups with electron withdrawing resonance effects such as nitro, amine, carbonyl groups and electron donating groups such as methylene.

Interactions on the hinge zone of compound 1 (A) and compounds 2-4 aligned (B), from Table 1.
In Figure 2A we can see the hydrogen bonds on the hinge zone to compound 1 (Table 1). This compound has two interactions on the hinge zone with the residues Glu96, Val98 of 2.29 Å and 2.54 Å, respectively. Another hydrogen bond can be formed with the residue Asn104 of 1.98 Å. Compounds 2, 3 and 4, see Figure 2B, have differences only in the substituent group, in the case of 3, this is a pyrrole ring and in 4, is the dimethyl-amine group. In this series, the most active compound is 2, with pIC50 -1.839. These compounds show two hydrogen bonds on the hinge zone with residues Glu96 and Val98 of 1.31 Å and 2.30 Å to 2, 1.71 Å and 2.35 Å to 3 and 1.81 Å and 2.21 Å to 4. Another hydrogen bond can also be formed with the residue Gly145 of 2.22 Å to 2, 1.81 Å to 3 and 1.95 Å to 4. From these results, we can see the key factor to the stabilization of the active site in this ligand set, with the main characteristic of one H-bond acceptor and H-bond donor in this zone.
Figure 3 (left) and Figure 4 show, respectively, the atomic charges calculated by the CHelpG population and electrostatic potential analyses of compound 1 in some areas of special interest. The net charges obtained for H22, O24, O30 and O31 are consistent with the formation of hydrogen bonds shown in Figure 2A. The electrostatic potential surfaces shown in Figure 4 also justify the formation of hydrogen bonds with Glu96, Val98 and Asn104. Figure 3 (right) and Figure 5 show, respectively, the atomic charges and electrostatic potential of compound 4. The atomic charges of H27, H42 and H15 justify very well the formation of hydrogen bonds with Glu96, Val98 and Asn104 that can be seen in Figure 2B. An equivalent conclusion can be found from the electrostatic potential surfaces shown in Figure 5.



The compounds to Pkn B have groups with electron withdrawing resonance effects, such as carbonyl, hydroxyl, amine and methoxy groups, with big rings central to its stabilization on the active site. Figure 6 shows the docking results of the Pkn B inhibitors from Table 2–Table 5, grouped by series, taking into account their functional groups and structural similarities. Figure 6A shows compound 5, which has interactions in the hinge zone with the residues Glu93 (2.63Å) and Val95 (-CO: 2.46Å, -NH: 1.99Å, 2.16Å), see Table 7. Compound 5 has dual effects, interacting with Pkn B (pIC50: -0.585) and Pkn G (pIC50: 1.301), see Table 2. Figure 6B shows the interaction with compound 6, which has interactions with the residue Val95 (-CO: 1.31Å, -NH: 2.48Å). In these interactions the -NH is H-bond donor and the –CO is H-bond acceptor; these interactions can be related to the pIC50 of 0.569 for this compound.

Figure 6C shows the interaction in the hinge zone to of compounds 7, 8 and 9. These compounds have two interactions in the hinge zone with the residue Val95. The most active compound of this series is 8 (pIC50: 0.585) with its interactions with the residue Val95 (-CO: 1.80Å, -NH: 2.15Å), see Table 7. Figure 6D shows the interactions of the compound 10; this compound has two interactions in the hinge zone with the residue Val95 (-NH: 2.33Å, -C=O: 2.09Å). Another H-bond happens with the residues Asp96 (1.71Å) Asp102 (-C=O: 2.42Å, -OH: 2.10Å) and Lys140 (2.01Å). The interactions with the two guanidine groups are through the aspartates. This is very interesting because these kinds of interactions could differentiate one kinase from another. Figure 6E shows the interactions in the hinge zone of the compounds 11, 12, 17, 18, 19, 20 and 23–53. This series of compounds have three H-bonds in the hinge zone with the residues Glu93 and Val95. The most active compound from this series is 52 (pIC50: 1.638). It has interactions with residues Glu93 (2.21Å) and Val95 (-CO: 2.33Å, -NH: 1.85Å), see Table 7. In this series there are two –CO H bond acceptors and one -NH H bond acceptor. Figure 6F shows the interactions in the hinge zone of the compounds 13 and 22; these compounds have two interactions in the hinge zone with the residue Val95 (-CO: 2.39Å, -NH: 2.42Å for 13 and -CO: 2.27Å, -NH: 1.90Å for 22), see Table 7. The most active compound is 22 (pIC50: 1.187). Finally, Figure 6G shows the interactions in the hinge zone of the compounds 14, 15, 16, 21 and 54–57. This series of compounds also has three interactions in the hinge zone with the residues Glu93 and Val95. The most active compound in this series is 57 (pIC50: 1.398), see Table 5.
Taking in account the results from Table 7, the key factor to the stabilization of the active site of these ligands is one or two –CO (H bond acceptor) and one -NH (H bond acceptor) groups in this zone.
Figure 7 shows the atomic charges calculated with the CHelpG population analysis (upper left) and the electrostatic potential (upper right and down) of compound 5 in some areas of special interest. The net charges obtained for H11, O13, H21 and H22 are consistent with the formation of hydrogen bonds shown in Figure 6A. The electrostatic potential of surfaces shown in Figure 7 also justify the formation of hydrogen bonds with Glu93 and Val95.

Figure 8 shows the atomic charges calculated by CHelpG population analysis (left) and the electrostatic potential (right) of compound 6 in some areas of special interest. The net charges obtained for H40 and O41 are consistent with the formation of hydrogen bonds shown in Figure 6B. The electrostatic potential of surfaces shown in Figure 8 also justify the formation of hydrogen bonds with Val95.

Figure 9 shows the atomic charges calculated by CHelpG population analysis (left) and the electrostatic potential (right) of compound 8 in some areas of special interest. The net charges obtained for N9 and H13 are consistent with the formation of hydrogen bonds shown in Figure 6C. The electrostatic potential of surfaces shown in Figure 9 also justify the formation of hydrogen bonds with Val95.

Figure 10 shows the atomic charges calculated by CHelpG population analysis (left) and the electrostatic potential (upper right and lower) of compound 10 in some areas of special interest. The net charges obtained for O29, N31, N16, H64 and H76 are consistent with the formation of hydrogen bonds shown in Figure 6D. The electrostatic potential of surfaces shown in Figure 10 also justify the formation of hydrogen bonds with Val95 (O29 and N31), Lys140 (N16) and Asp102 (H64 and H76).

Figure 11 shows the electrostatic potential generated by compound 13 in some areas of special interest. The electrostatic potential of surfaces shown in Figure 11 are consistent with the interactions shown in Figure 6F and justify the formation of hydrogen bonds of H34 and N36 with Val95.

Figure 12 shows the atomic charges calculated with the CHelpG population analysis (left) and the electrostatic potential (right) of compound 15. The net charges obtained for H31, H38 and N39 are consistent with the formation of hydrogen bonds shown in Figure 3G. The electrostatic potential of surfaces shown in Figure 12 also justify the formation of hydrogen bonds with Glu93 (H38) and Val95 (H31 and N39).

The Pkn G inhibitors from Table 6 have a common structure and three interactions in the hinge zone with the residues Glu233 and Val 235 (see Figure 13).

The most active compound of this series is 66 (pIC50: 2.000). This compound has interactions with the residue Glu93 (2.59Å) and Val95 (-CO: 2.24Å, -NH: 2.17Å), see Table 8. Other compounds with good reactivity are 62, 63C1 and 63C2 (pIC50: 1.699). These compounds have interactions with the residues Glu93 (2.18Å), Val95 (-CO: 2.56Å, -NH:1.78 Å); Glu93 (2.36Å), Val95 (-CO: 2.18Å, -NH:1.90 Å) and Glu93 (2.10Å), Val95 (-CO: 2.22Å, -NH:1.89 Å), respectively. In these ligands the key factors are two –CO (H bond acceptor) and one -NH (H bond acceptor) groups for stabilizing the ATP active site.
| Compound | Hinge zone | Others (–H bonds) | ||
|---|---|---|---|---|
| Glu233 | Val235 (-C=O) | Val235 (-NH) | ||
| 58 | 1.99 | 2.37 | 1.80 | - |
| 59 | 2.27 | 2.74 | 2.36 | - |
| 60 | 2.07 | 2.51 | 1.88 | - |
| 61 | 2.57 | 2.57 | 2.18 | - |
| 62 | 2.18 | 2.56 | 1.78 | - |
| 63C1a | 2.36 | 2.18 | 1.90 | Lys181:2.33 |
| 64C2a | 2.10 | 2.22 | 1.89 | Asp293:1.68 |
| 65 | 2.69 | 2.64 | 1.93 | Lys181:2.31 |
| 66 | 2.59 | 2.24 | 2.17 | Lys181:2.16 |
| 67C1a | 2.47 | 2.61 | 1.93 | Asp293:2.00 |
| 68C2a | 2.28 | - | 1.98 | Asp293:1.81 |
| 69 | 2.43 | 2.67 | 1.87 | - |
| 70 | 1.96 | 2.60 | 1.73 | - |
| 71 | 2.60 | 2.56 | 1.79 | - |
| 72 | 2.00 | 2.49 | 1.80 | - |
| 73 | 2.27 | 2.06 | 1.94 | - |
| 74 | 2.43 | 2.74 | 2.24 | - |
| 75 | 2.07 | 2.60 | 1.80 | - |
| 76 | 2.47 | - | 2.56 | - |
| 77 | 2.03 | 2.55 | 1.82 | - |
Figure 14 shows the atomic charges calculated by CHelpG population analysis (left) and the electrostatic potential (right) of compound 58. The net charges obtained for H16, O11 and H14 are consistent with the formation of the hydrogen bonds shown in Figure 13. The electrostatic potential of surfaces shown in Figure 14 also justify the formation of hydrogen bonds with Glu233 (H16) and Val235 (O11 and H14).

The MQS analysis was conducted to understand the correlation between the ATP ligands associated to each kinase studied and the stabilization process in the active site. This correlation allows postulation of the structural models of the ATP complexes.
For the Pkn A ligands (see Figure 15), the correlation between the steric and electronic effects is R2 =0,9487 using the coulomb and overlap operators to determine electronic and steric effects, respectively. This correlation shows the nature of the stabilization of these ligands on the active site and its non-covalent character.

Figure 16 shows the correlation of Pkn B ligands (see Table 2–Table 5). This correlation shows the electronic and steric effects along the ligand set associated with the active site. The high values in the Carbó indexes give an idea about the ATP complex generated and stabilized.

In Figure 17 is shown the correlation between the steric and electronic effects associated with the ATP complex of Pkn G (see Table 6). This complex has a correlation of R2=0,7357. In this complex, the steric and electronic effects also are very relevant and the stabilization in the active site has a high non-covalent character.

In Figure 15–Figure 17 a strong correlation between the two operators used to determine electronic and steric effects is shown. These effects can be related to the stabilization process and non-covalent interactions. In this way, the quantum similarity analysis allows us study the ATP complex and understand the steric and electronic relationship along the ligand set.
Inadequate dosing and incomplete treatment regimens, coupled with the ability of the tuberculosis bacilli to cause latent infections that are tolerant of currently used drugs, have fueled the rise of multi-drug resistant tuberculosis. To deal with this problem, there is currently an increased interest in kinase inhibitors with the main aim of proposing new anti-tuberculosis drugs. For these reasons, in this work we conducted a study of the crystalized STPKs Pkn A, B1, B2, B3 and G. The conformational analysis shows the DFG-in state of Pkn B, G, and DFG-out state of Pkn A. Methionine is the gatekeeper residue in all the kinases studied.
To understand the interactions of the active site of the protein kinases from Mycobacterium tuberculosis, a docking study using the protein kinases Pkn A, B and G was conducted. In these protein kinases, the C-term domain is extracellular and N-term is cytosolic. The cytosolic domain is almost the same for all the kinases and the extracellular domain is different. Ligands bind to the kinase through the formation of two to three hydrogen bonds with the backbone residues of the hinge region connecting the kinase N- and C-terminal loops. The residues with hydrogen bonds on the hinge zone of Pkn A are GLU96 and VAL 98, of Pkn B are GLU 93 and VAL 95, and finally of Pkn G are GLU233 and VAL235. From these results, it is possible to see that the interactions of the hinge zone are characterized by the key factor of one or two H-bonds acceptors and one H-bond donor in the ligands of this zone. On the other hand, the DFT calculations performed with some of the most representative compounds have supported the previous conclusions. Finally, the quantum similarity analysis shows good correlation between the steric and electronic effects in each ATP complex. In this sense, the outcomes presented in this work can be used to develop new protein substrates of the STPKs and find new tuberculosis treatments.
All data underlying the results are available as part of the article and no additional source data are required.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)