Keywords
acute polymyositis, case report, hemophagocytic lymphohistiocytosis, HLH
acute polymyositis, case report, hemophagocytic lymphohistiocytosis, HLH
Hemophagocytic lymphohistiocytosis (HLH) is a rare immunological syndrome characterized by an uncontrolled hyperinflammatory response coupled with excessive lymphocyte and macrophage activation.1 People with this condition have a substantial risk of death and disability because of its severity and difficulty to diagnose. HLH can be grouped into two main types: primary and secondary, each of which has its own subtypes. Primary or familial HLH is an autosomal recessive inheritance often manifesting in infants and children. The main pathogenesis lies in genetic mutations or variations impacting the cytotoxic functions of natural killer cell (NK cell) and cytotoxic T cells.2 On the other hand, secondary HLH usually affects adolescents and adults;there are no known underlying genetic defects, and it rather occurs as a consequence of infections, malignancies, autoimmune diseases, among others.3,4 Rheumatological diseases have been linked to a significant number of cases of HLH around the world, with systemic lupus erythematosus (SLE) and systemic juvenile idiopathic arthritis (sJIA) being the most common; but very few cases have been reported with polymyositis.5 Our case report aims to highlight a rare and unique association between HLH and polymyositis so that the disease is identified and treated early and effectively to assure long-term survival.
A 14 year-old Bengali male was admitted to Chattogram Medical College Hospital with a high-grade fever for 25 days, generalized severe body aches, multiple large joint pain in the lower limbs for 20 days, ulceration at lips for three days and loss of bowel and bladder control for one day. Before being admitted to our hospital, the patient was taken to a local hospital. He was diagnosed with enteric fever and was treated with intravenous (IV) third-generation cephalosporin (ceftriaxone) but didn’t improve clinically and was referred to a tertiary care hospital. The patient was non-diabetic and normotensive, and had a previous history of asthma.
On examination, his temperature was 102°F (38.9°C), pulse 150 bpm, and blood pressure 80/60 mmHg. He had tenderness in both knee joints, proximal myopathy of both lower limbs, and several sensory losses.
His initial investigation revealed bicytopenia with elevated inflammatory markers, i.e., Erythrocyte sedimentation rate (ESR), C-reactive protein, and serum ferritin. Patient showed altered liver and renal function test; with elevated serum alanine aminotransferase (s. ALT), decreased serum albumin, decreased serum fibrinogen, elevated serum bilirubin, and elevated serum creatinine level. Moreover, serum creatine phosphokinase (s. CPK), serum lactate dehydrogenase (LDH), serum triglyceride, and serum d-dimer were found abnormally high. Serum electrolytes revealed hyponatremia (Table 1).
Ultrasonography of the whole abdomen indicated renal parenchymal disease, pleural effusion, and mild splenomegaly (10.7×5.1 cm) (Figure 1). Peripheral blood film revealed microcytic hypochromic anemia with neutrophilic leukocytosis with thrombocytopenia; bone marrow examination presented reactive marrow with erythroid hyperplasia with an increased number of lymphocytes. Investigations for screening autoantibodies, i.e., perinuclear anti-neutrophil cytoplasmic antibodies, anti-neutrophil cytoplasmic antibodies, rheumatoid factor, Antistreptolysin O titer, direct Coombs’s test, anti dsDNA, antinuclear antibodies, anti-cyclic citrullinated peptide antibody tests were negative. Sepsis screening for hepatitis B, C, human immunodeficiency virus (HIV), malaria, salmonella, rickettsia, and brucella was also negative. CSF study revealed no significant abnormalities, and a magnetic resonance imaging (MRI) of the Lumbosacral spine showed no significant finding.
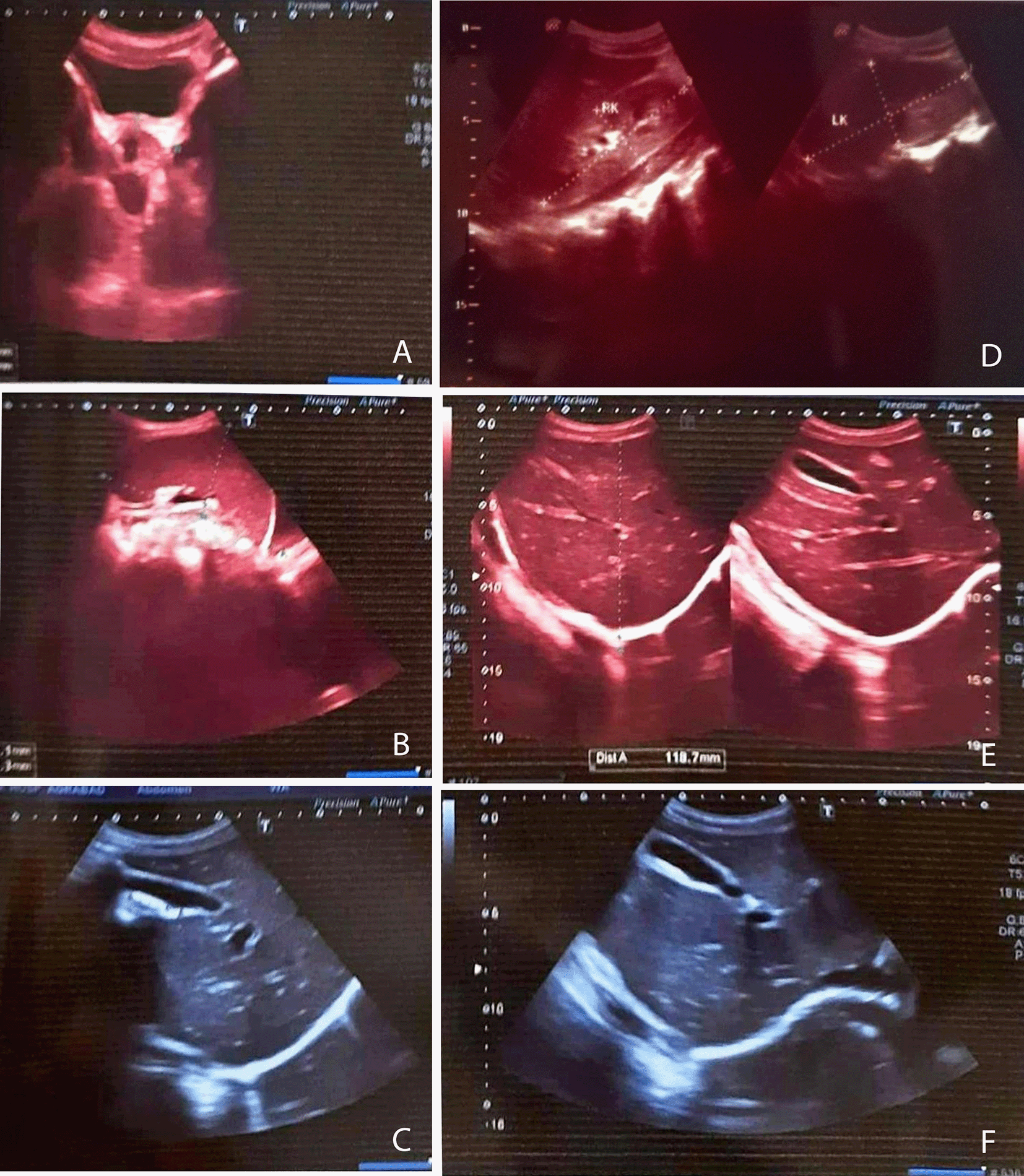
A. Urinary Bladder is well filled with normal wall thickness. Prostate is normal in size with intact capsule. B. Spleen was mildly enlarged in size (10.7x5.1 cm). C. Liver is normal in size measuring about 11.4 cm at the level of right anterior mid clavicular line (normal up to 14 cm) & normal parenchymal echotexture all over. D. Both kidneys were swollen, parenchymal echogenicity was increased, and corticomedullary differentiation was altered. E, F: Gall Bladder is well outlined, normal in size and wall thickness is within normal limit. Common bile duct and intrahepatic biliary channels are not dilated. Visible part of pancreas appears normal.
By the judgment of clinical presentation and investigation, we diagnosed the case as HLH syndrome with polymyositis. The patient was treated with IV hydrocortisone and dexamethasone. Clinical stability was achieved with gradual improvement of initial symptoms and biochemical markers. ESR and s. ferritin were reduced and liver and renal function tests were improved. Furthermore, s. CPK was also reduced (Table 1). The patient was discharged with oral steroids at a tapering dose and advised for regular follow-up with no clinical relapse that needed hospital admission.
In a normal immune response, natural killer cells (NK cells) and cytotoxic lymphocytes (CTL) suppress macrophages and CD8 cells to attenuate the immune response. However, deficient NK cell activity and CTL fail to suppress the immune response and result in profuse activation and release of interferon-gamma and inflammatory cytokines, causing organ damage. This pathogenesis plays a central role in HLH.6–8 Primary HLH is due to mutations in different genes such as PRF1, UNC13D, STX11, RAB27A, LYST, AP3B1, and SH2D1A that regulate perforin expression in immune cells and regulates synthesis, maturation, and release of cytotoxic and cytolytic granules in lymphocytes.9 Most of these are inherited as autosomal recessive form secondary associated with infections, autoimmune or malignant disorders.8,10,11
According to the HLH-2004 trial criteria,12 diagnosis of HLH is established if one of either (1) or (2) below (Table 2) is fulfilled.
The present case fulfilled five out of eight diagnostic criteria of HLH; fever, splenomegaly, bicytopenia, hypertriglyceridemia, and hyperferritinemia. Furthermore, hepatopathy with altered liver function test, coagulopathy, raised d-dimer, hypoalbuminemia, and hyponatremia strengthen the diagnosis of HLH.6,13,14 Furthermore, markedly elevated skeletal muscle-related enzymes (i.e., s. CPK, s. LDH, and s. ALT) in association with symmetrical proximal muscle weakness, severe myalgia, and absence of rash indicate acute polymyositis.
The collapse of immunological tolerance against muscle antigen is the fundamental mechanism of polymyositis.15 Hence, the combination of the two autoimmune diseases of seemingly unrelated systems in this patient could be a reflection of the widespread impairment of immune functioning in the patient as a whole. HLH has been reported in systemic lupus erythematosus, Still’s disease, rheumatoid arthritis, systemic juvenile arthritis, dermatomyositis, Kawasaki disease, systemic sclerosis, Bechet’s disease, polyarteritis nodosa, ankylosing spondylitis, mixed connective tissue disease, sarcoidosis, Sjogren’s syndrome, Wegener’s granulomatosis in previous case reports but very few care reports were indexed with polymyositis.16,17
There are exceedingly few reported occurrences of HLH linked with acute PM. This article seeks to add to the existing knowledge by describing the characteristics of a probable HLH presentation with PM. HLH with PM should be suspected in a patient with unexplained fever, cytopenia, hepatitis, proximal muscle weakness, and myalgia. The diagnosis is difficult because patients may appear with symptoms that are indistinguishable from those of sepsis or multiple organ failure syndrome. Priority should be given to quick workup using complete blood count (CBC), inflammatory markers, liver function test, triglycerides, and bone marrow examination. Early diagnosis and prompt treatment are the key to improving survival in this patient group.
The mother of the patient (since the patient was a minor) provided written informed consent for this report. Since this is a case report that maintains the anonymity of the patient, ethical clearance from our Institutional Review Board was not necessary. This report was prepared in accordance with the CARE guidelines.18
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)