Keywords
Parkinson drugs; Activity Cliffs; Chemoinformatics; Dual Activity Cliffs; Polypharmacology; Structure-Activity Relationships.
This article is included in the Cheminformatics gateway.
Parkinson drugs; Activity Cliffs; Chemoinformatics; Dual Activity Cliffs; Polypharmacology; Structure-Activity Relationships.
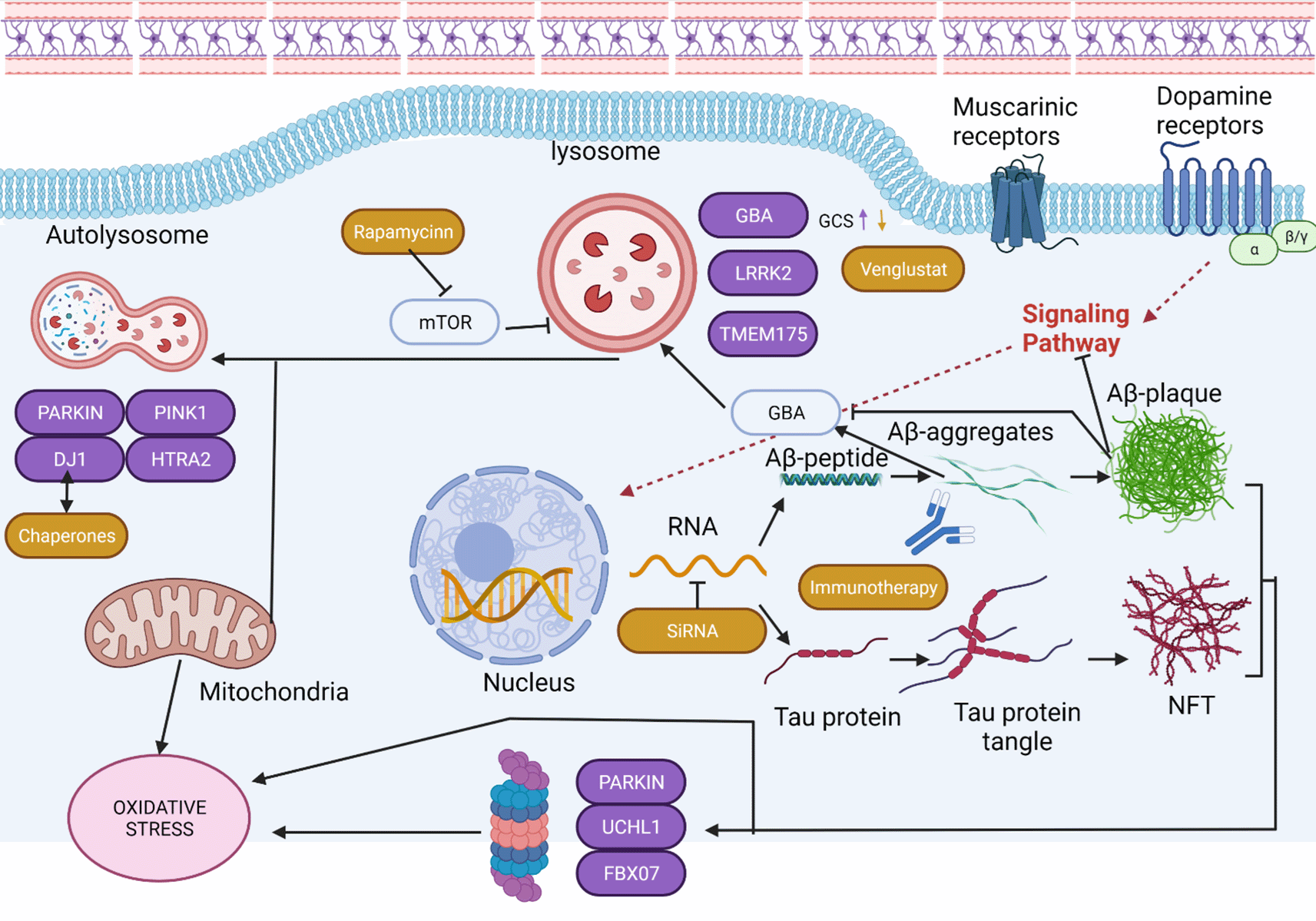
Parkinson’s disease (PD) is a complex and multifactorial neurodegenerative disease (Ontology: DOID:14330; OMIM: 168600).1 The underlying aspects of the neurodegenerative process have been unraveled and are becoming known, while treatments that can modify this process are still in the experimental stage. Currently, PD has a symptomatic treatment derived from the gradual loss of neurons in the substantia nigra, which contributes to motor and non-motor symptoms. At the onset of the disease, motor symptoms respond predominantly to levodopa treatment. However, the symptoms of chronic PD tend not to respond satisfactorily to levodopa treatment, which is partly explained by the fact that PD is considered a complex disease combining genetic and environmental factors, in which genes can either generate the disease on their own or be part of the risk factors. Parkinson Disease are associated with five targets, one of this is an immuno-relevant target. In Figure 1 shows the protein targets that have been associated with various stages of PD. It should be noted that in the Synuclein Alpha (SNCA) pathway siRNAs have been described as negative regulators. Likewise, some small molecules such as Nilotibib are immunoregulators.2
The PD patients have an insidious quality of life, which is stressful due to treatments based solely on symptomatic features. As the world’s population continues to age, the prevalence of PD is expected to double in some age groups, placing a considerable burden on healthcare systems. Fortunately, the rise of rational polypharmacological approaches is increasing, in particular for the treatment of multifactorial and complex diseases, such as neurodegenerative diseases. PD is an ideal model for a polypharmacological approach due to its clinical heterogeneity. Indeed, the search for specific targets has been the basis of pharmacology for many years. With the advent of systems pharmacology, a quantitative area based on computational methods it has become possible to develop compounds directed to more than one target. In this sense, it has been possible to take data from fields such as clinical neurophysiology and neuropharmacology, and organize them with high-throughput screening methods, such as ligand-based virtual screening, or structure-based virtual screening. Thus, PD compounds represent a promising approach to shed light on the etiology and treatment of complex neurodegenerative diseases such as PD. There are few candidate polypharmacological compounds targeting dopaminergic neurons,3–5 although none of them are in clinical development yet. For this reason, we are interested in contributing to show polypharmacological opportunities in PD.
Chemoinformatics allows systematic studies of chemical structure discernment and comparison of chemical structures present in compound databases. The relationships that can be established through informatics strategies reveal pharmacological or polypharmacological compounds with PD-related endpoints as a common factor. Polypharmacology is the binding of drugs to multiple targets, the main one being the search for lead structures that interact with several biological targets related to a specific pathological entity; not others that may produce unwanted adverse effects.6 Therefore, the goal of this paper is to identify compounds with over one receptor target related to PD.
The compound dataset was constructed in three steps: (1) An initial dataset with 15,119 compounds, tested against different human (Homo sapiens), rat (Rattus norvergicus), and mouse (Mus musculus) endpoints. In the same way, datasets related to the development of PD (reported in the ChEMBL V.30 database7), (2) Then, duplicated structures were removed, yielding a dataset with 5,992 unique compounds (see Table S1 in the Supplementary Material). In the case of duplicated compounds, those with the highest reported activity (< IC50) were retained. For this analysis, compounds with dose-response values equal to or lower than 10 μM were considered “active”, and compounds with higher values were considered “inactive”. It should be noted that a value of 10 μM has been used as a general threshold to define active/inactive molecules in other large-scale studies8; (3) 1,562 compounds were considered active for almost one set of the data (see Table S2 in the Supplementary Material). All compounds have been associated with the IC50 values indicated for each data set. SMILES representation of structures and the pIC50 (-log IC50) values are summarized in Table S1 of the Supplementary Materials. In general, the range of activity values for each set is similar, which facilitates cross-comparisons of the activity landscape (vide infra).9
Activity cliffs (AC) represent pairs of compounds with high structural similarity but with an unexpected change in their biological activity. The Structural Activity Landscape Index (SALI) value has been commonly used in the systematic identification of AC.9
AC has been optimized by identifying chemical shifts related to enhanced biological activity against two data sets simultaneously (Dual activity cliffs and D-AC). Here, the dual activity difference (DAD) map is a tool for structure-activity relationship (SAR) analysis of data sets of compounds tested against two molecular targets. DAD maps are based on the activity landscape concept and are suitable for the fast identification of “selective switches”, defined as compounds with structural changes, that completely reverse the selectivity towards two different biological targets.10 DAD maps are based on systematic pairwise comparisons of compounds in a dataset, comparing similarity (using the extended connectivity fingerprint of radius 4 - ECFP4 - fingerprint, and the Tanimoto coefficient), and differences in activity (pIC50) against two targets (simultaneously) for each compound pair.
This work describes an extension of the DAD map that allows the rapid identification of D-ACs. The newly proposed 3D Dual map includes a Z-axis that represents the Dual Structural Activity Landscape Index (D-SALI). which Is calculated with the equation:
where:A1 = Values of activity against the first target.
A2 = Values of activity against the second target.
i = The first compound on the pair analyzed.
j = The second compound on the pair analyzed.
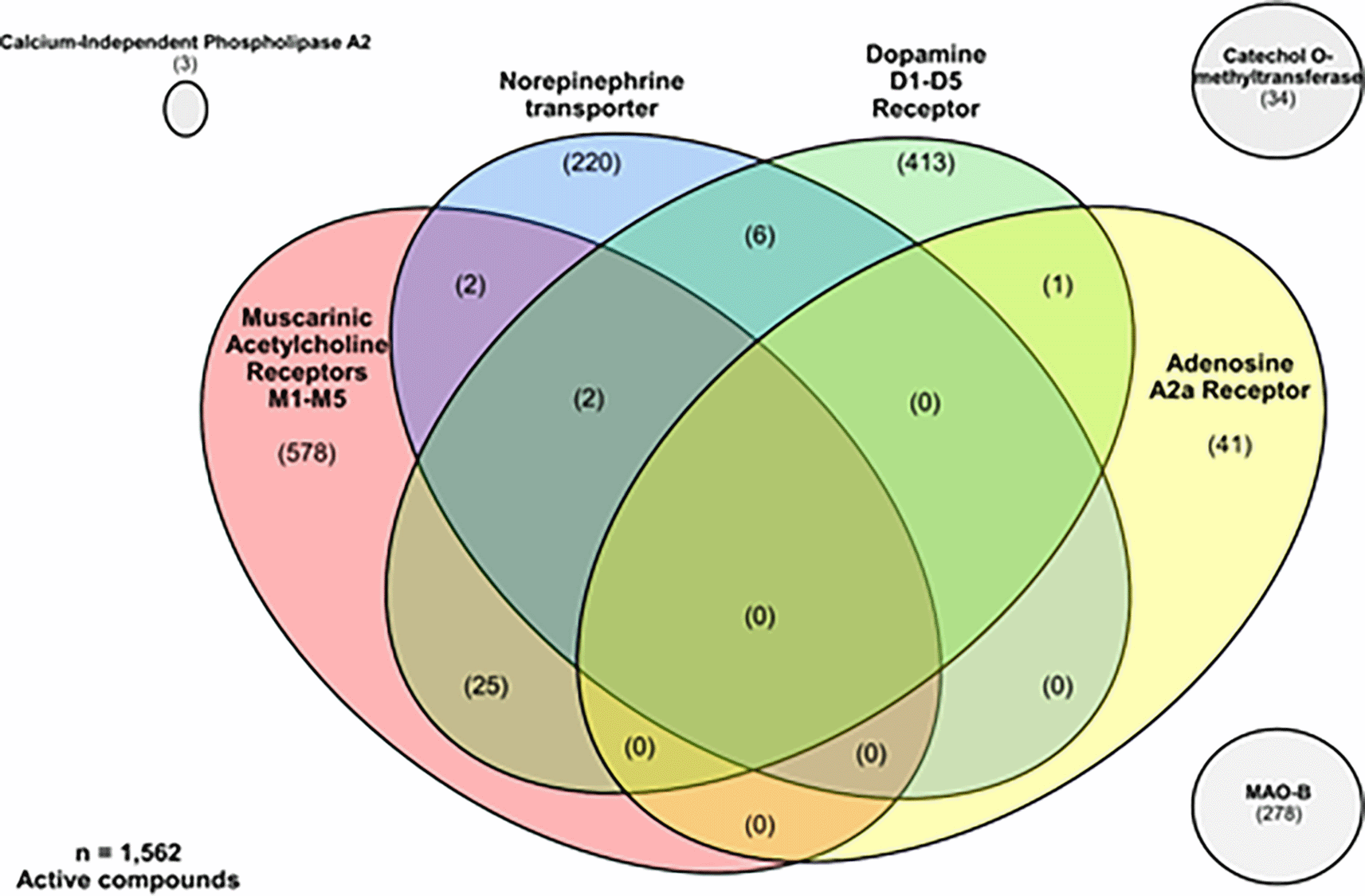
In our study we selected 1,562 active compounds (IC50 equal to or lower than 10 μM) reported against almost one target related to PD, shown in Figure 2. It is relevant that 36 of the compounds have a polypharmacological activity against over two different target families (Table S1). Of these multi-target compounds, only two exhibit activities against different target families related to PD (e.g., muscarinic acetylcholine receptors, norepinephrine transporter, and dopamine receptors). Additionally, strategic search reveals promising chemical dual-entities. For example, a compound with activity against adenosine A2a and dopamine D1 receptors, six compounds with activity against norepinephrine transporter and dopamine receptors (D1-D5), and two compounds with dual activity against norepinephrine transporter and muscarinic acetylcholine receptors (M1-M5). Interestingly, 25 compounds with dual activity against dopamine receptors and muscarinic acetylcholine receptors has been identified. The chemical structures and activity profile of representative polypharmacological compounds were shown in Figures 3 and 4.
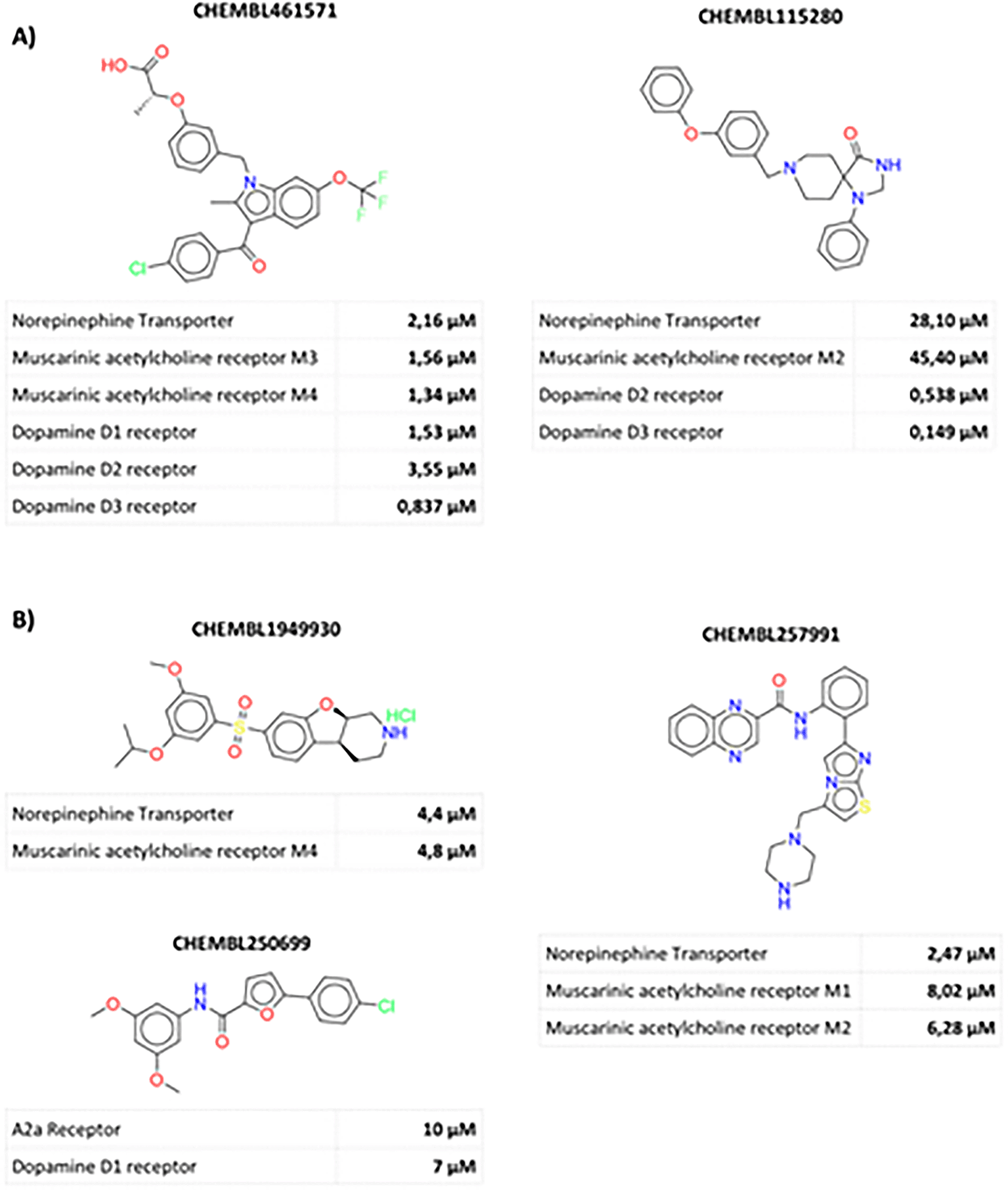
A) Representative Poly-active compounds. B) Dual compounds related with dopamine and muscarinic acetylcholine receptors.

Examples of the most studied targets related to PD, are the dopamine and muscarinic acetylcholine receptors. These targets are associated with multiple biological functions in the CNS (e.g., dopaminergic denervation) and diseases (e.g., Huntington’s disease, Alzheimer’s disease, PD, etc.) For this reason, the pharmacological irruption of these targets is one of the most important topics in drug discovery and design. Particularly, in PD, the dual inhibitions (dopamine and muscarinic acetylcholine receptors) have been associated with their symptom improvement,20 which suggests that is a promising strategic the explicit design of compounds with dual activity. According to this idea, Figure 4 shows an overview of the most potent active compounds (reported on ChEMBL V.30) against dopamine (D1-D5) and muscarinic acetylcholine (M1-M5) receptors.
As been illustrated in Figure 4, the scaffold N-(4-(4-(benzo [d]isothiazol-3-yl)piperazin-1-yl)butyl) acetamide is a representative structure of the most potent dual inhibitors (dopamine and muscarinic receptors). This scaffold is present in six of the ten most potent dual compounds (2,4,5,6,9 and 10). The associated activity of this scaffold is only surpassed by compound 1 (CHEMBL4128926) which has higher activity against both targets (especially, is more potent against muscarinic receptors). Additionally, CHEMBL4128926 has been associated with other CNS targets (e.g., GABA-gated chloride channel, 6 and 2 serotonin receptors, Mu and Kappa opioid receptors, and neurokinin receptors 3).21 Interestingly, their pharmacokinetic profile reveals that have a desirable oral bioavailability, distribution volume, and half-life on in vivo (Rattus norvergicus) models.22,23 In contrast, compound 2 (CHEMBL343838), which contains the representative scaffold N-(4-(4-(benzo [d]isothiazol-3-yl)piperazin-1-yl)butyl) acetamide, has been associated with the inhibition of the serotonin 1a and 2 receptors, and it has an acceptable ADMET profile.24
The identification of AC is a crucial step in the complete understanding of Structure–Multiple Activity Relationships (SMARt).25 For compound datasets with measured experimental activity. Small changes in the chirality, chemical substituents, and scaffold are the common reason to generate cases of AC. Figure 5 shows four representative dual AC cases against dopamine and muscarinic acetylcholine receptors.
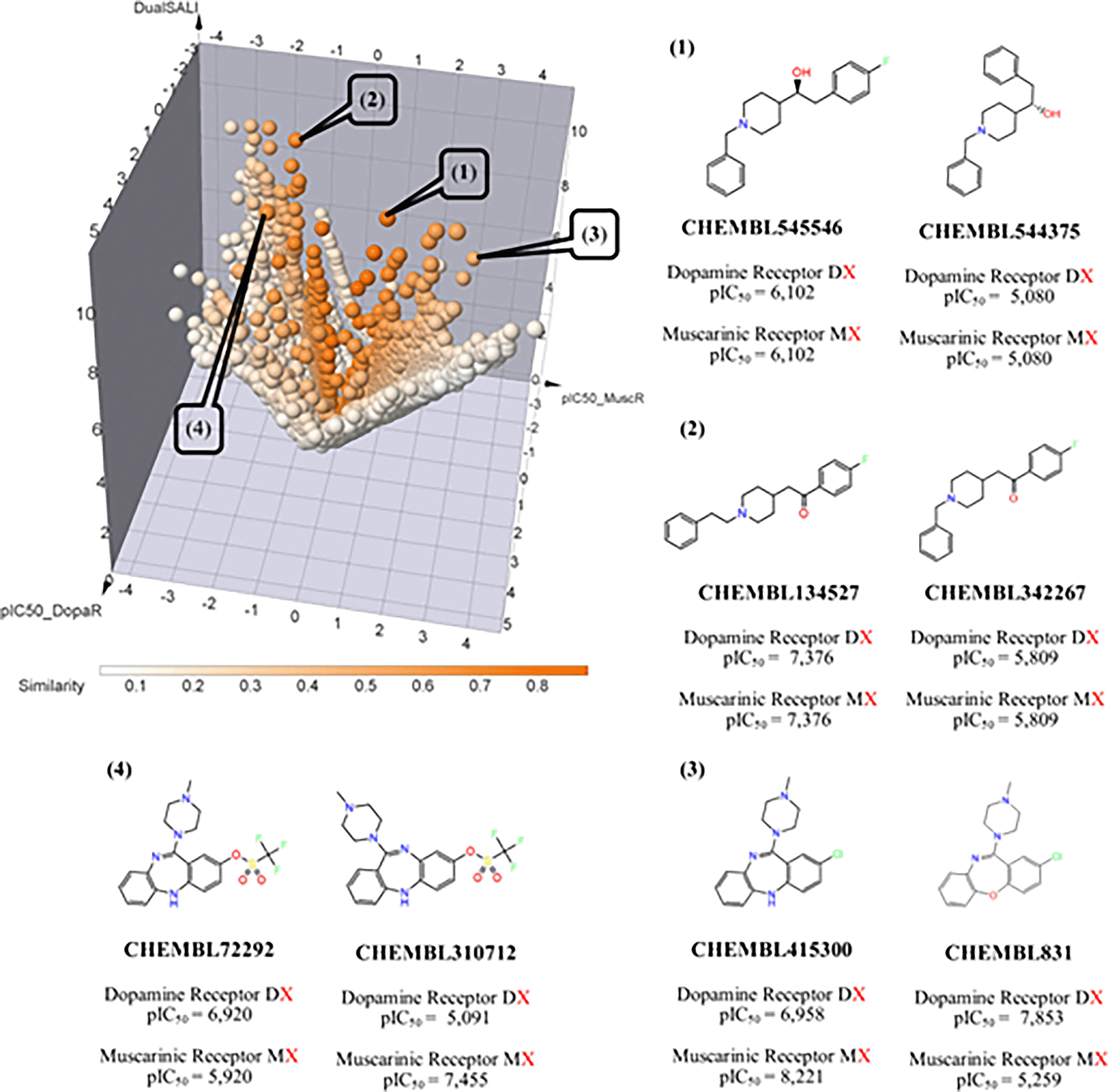
Example (1) refers to the pair of compounds CHEMBL545546 against CHEMBL544375 (see Figure 5) with a chirality change on the carbon bonding with the -OH group on the structures. This chiral change reduces the activity against both targets in ~1 logarithmic unit. This observation suggests that dopamine and muscarinic acetylcholine receptors could be stereoselective. Besides, the example (2) refers that the elongation of the structure is another key factor in the design of new dual active compounds. For example, CHEMBL134527 (larger) in contrast to CHEMBL342267 (shorter) increases the activity against both targets in ~1 logarithmic unit.
In contrast with pair of compounds 1 and 2, the example (3) (CHEMBL72292 and CHEMBL310712) illustrates an example of an isomeric change that increases the activity against dopamine receptors in ~2 logarithmic units but decreases the activity against muscarinic acetylcholine receptors in ~2 logarithmic unit. Finally, study case (4) exhibits a bidirectional activity profile from the substitution of nitrogen by oxygen on the scaffold (i.e., the isomeric change decreases the activity against dopamine receptors in ~2 logarithmic units but increases the activity against muscarinic acetylcholine receptors in ~2 logarithmic units).
The examples described in Figure 4 are multi-target compounds with improved IC50s against dopaminergic and muscarinic receptors. In fact, cases (1) and (2) show that is possible to identify chemical changes associated with the activity improvement against multiple endpoints, but that also is possible to regulate the selectivity of these compounds without loss of potency against a specific endpoint (as shown in the case (3) and (4)).
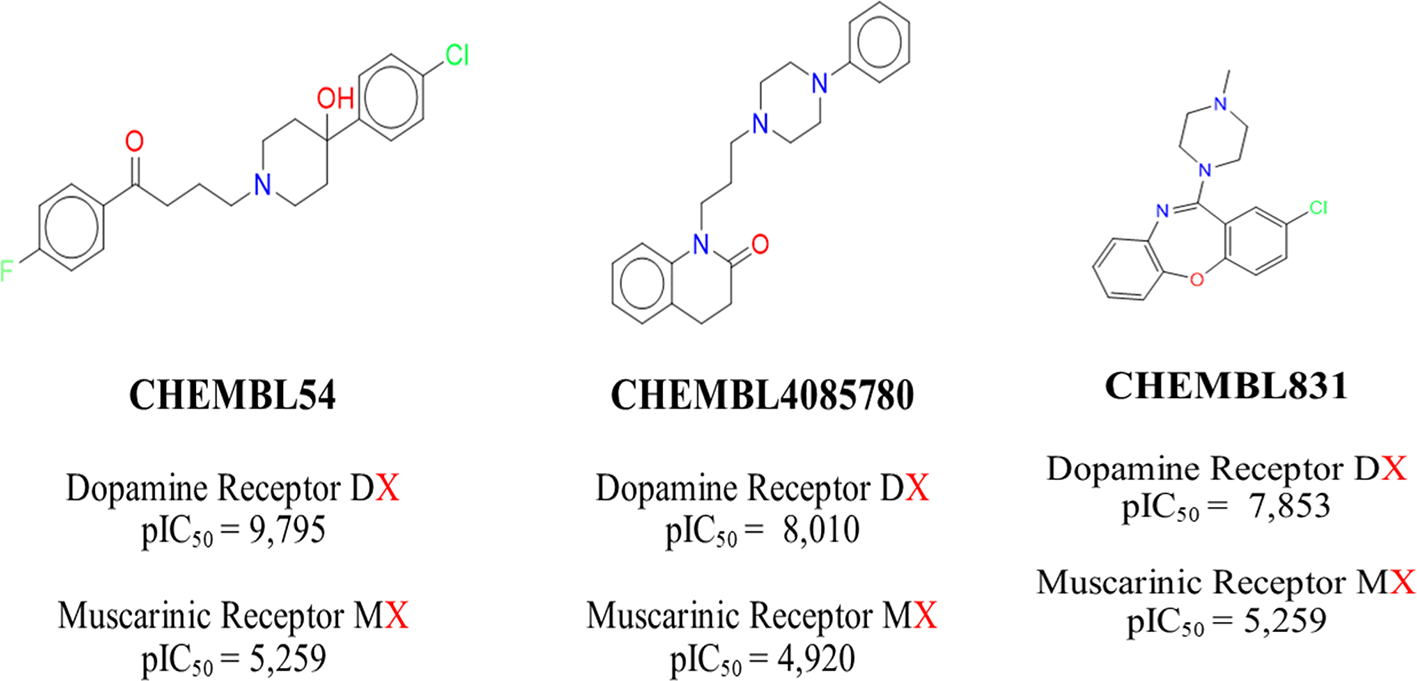
Interestingly, there are compounds with large activity differences (around 3 logarithmic units) with the dopamine and muscarinic acetylcholine receptors. Figure 6 shows examples of selective compounds. For example, we highlight the selectivity of CHEMBL54 (haloperidol), CHEMBL4085780, and CHEMBL831 against the dopamine receptors.
As discussed in the Introduction section, multi-target therapy offers new perspectives that could resolve different issues in relationship with single-target drug design. At same time, polypharmacology therapy offers the possibility to generate novel and refined approaches to address to complex diseases such as neurological diseases. In this context, the generation and optimization of polypharmacological agents, represents a new challenge in drug design.
This study is an overview of multitarget compounds reported in the literature with potential application against PD. The data was obtained from ChEMBL V.30 (the most recent version, at the time or writing). These data do not represent all the compounds reported in the public domain against the different related targets on PD. For example, there are other primary resources with reported activity data such as PubChem or Binding DB, as well as those of private companies,26 that have not been included in this study. For example, a recent case remarks the not exhaustive data exploration on the literature (related with PD), is the case of 9-deazaxanthine derivatives, that has been dual against A2A (antagonists) and MAO-B (inhibitors), reported with nanomolar activity.27 Furthermore, this study is limited by the targets selected (Figure 2) to explore. This is a key point in the design and optimization of new chemical entities against PD.
This study uncovers the possibility of optimizing the tested compounds (e.g., shown in Figures 3-6) to identify new chemical structures and features related to a polypharmacological or selective profile against different PD targets. This offers a possibility to identify compounds that act on different molecular levels (i.e., changing the signaling, gene expression, or physiological effects) on PD. In fact, the parallel modulation of different endpoints could contribute to reducing the necessary doses to generate a therapeutic effect, that at the same time contributes to reducing the associated side effects on in vivo models.
Currently, there are 15 original and 3 repurposing compounds that have been approved for clinical use and other 19 compounds continue to be tested in clinical trials phase 3 with a single drug target.28,29 This data remarks a key opportunity to change the paradigm of “one compound one target” in drug discovery, and that the “promiscuity” concept should not be associated (directly) with serious side effects.
This new drug design approach has been guided by computational methods that have contributed to reducing the gap in information related to the development of new poly-active compounds. For example, now machine and deep learning techniques contribute to predicting and identifying optimized compounds against two or more endpoints.30 However, this model has been constructed using classification, clustering, or regression models that have not been possible to identify small chemical changes related to unexpected activity changes (i.e., activity cliff, AC). Accordingly, Figure 5 shows a new “proof of concept” to explore and use the reported dual-activity data to identify D-AC, and using D-SALI values to identify the most prominent selective and dual compounds on a data set. In fact, a perspective is an adaptation of the D-SALI value to identify triple (or quadruple, quintuple, etc.) activity cliffs.
This study has illustrated the potent (<10 μM) poly-active compounds against different targets related to PD existence. However, same compounds have been associated with promiscuity against other central nervous system (CNS) targets. For example, CHEMBL461571 and CHEMBL115280 (Figure 3-A). They have also been associated with opioid and adrenergic receptor and dopamine transporter uptake, to name a few of these targets.11,12 CHEMBL250699 (Figure 3-B) has been associated with the irruption of serotonin receptors,13,14 CHEMBL257991 are other examples with associated activity against adrenergic receptors and sirtuins (epigenetic targets),15,16 and CHEMBL1949930 with activity against serotonin receptor and dopamine transporter.17 Interestingly, these results reveal the promiscuity of some compounds interacting with different targets in the CNS.
Despite the high CHEMBL115280 and CHEMBL250699 promiscuity, these compoundshave been associated with a promising bioavailability profile on in vivo models.18 Another excellent case, reflecting these compounds potential, is CHEMBL1949930, which exhibits low toxicity, brain permeability, and a good bioavailability profile.19 These examples show that not in all cases poly-activity is associated with toxicological or bioavailability issues.
In the last decade, techniques and tools have emerged to design dual compounds; this facilitates the design of multi-target polyactive drug candidates. It is now possible to optimize compounds for two end-targets at the same time, provided the data exist to process them. In the next section, we present a study case of 25 compounds, with dual activity against dopamine receptors (D1-D5) and muscarinic acetylcholine receptors (M1-M5), illustrated In Figure 3.
In this paper, identified 36 polypharmacological chemical scaffolds related to Parkinson’s disease. In this manner we demonstrate that design of polypharmacological drugs is an opportunity in Parkinson’s Disease treatment.
In recent years, the old concept that drug selectivity meant having a single drug target has been left behind. Mainly, in multifactorial diseases, which involve interactions between molecular, cellular, and physiological pathways. Thus, in neurodegenerative disorders, including PD, the polypharmacological design approach is a fertile field for the development of new therapeutic strategies.
Figshare. Supplementary material, DOI: https://doi.org/10.6084/m9.figshare.21096919.v1.31
This project contains the following data:
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)