Keywords
HIV-1, antiretroviral therapy, treatment experienced, switch study, virally suppressed, randomised trial, antiretroviral therapy - adult, clinical trials, developing countries
HIV-1, antiretroviral therapy, treatment experienced, switch study, virally suppressed, randomised trial, antiretroviral therapy - adult, clinical trials, developing countries
Until December 2018, the World Health Organization (WHO) recommended that people living with HIV-1 infection who developed treatment failure on a non-nucleoside reverse transcriptase inhibitor (NNRTI)-based first line regimen should switch to a second-line regimen containing a ritonavir-boosted protease inhibitors (PI/r) plus two nucleoside reverse transcriptase inhibitors (NRTIs).1 The WHO now recommends the use of dolutegravir (DTG) as a second-line regimen for those who are integrase inhibitor (INSTI) naïve,2 however many countries, including Kenya, have been using PI/r-based second-line regimens for over a decade3 and almost all second-line patients in Kenya are currently on PI/r-based regimens, with approximately 75% of these patients virally suppressed.4
A second-line regimen consisting of DTG plus two NRTIs was shown to be superior to LPV/r plus two NRTIs after failing a first line regimen of NNRTI plus two NRTIs in the DAWNING trial.5 Results from the NADIA study demonstrated that a second-line regimen of DTG plus two NRTIs was non-inferior to DRV/r plus two NRTIs even among a population with extensive NRTI resistance.6
Two clinical trials have demonstrated that people without prior treatment failure who are virally suppressed on a PI/r-based regimen can safely switch to a DTG-based regimen, with non-inferior virological outcomes7,8 and improved patient satisfaction8 and lipid parameters DTG.7
No study to date has evaluated a switch strategy from PI/r to DTG for virally suppressed treatment-experienced patients who have failed a prior first line regimen consisting of a NNRTI plus NRTIs. If such a strategy proves safe and effective it will have major implications for the current regimens’ distribution in Kenya and other regions, allowing for almost 75% of current second-line patients to transition from PI/r-based regimens to a DTG-based regimen with lower cost, improved tolerability, decreased risk of toxicity, reduced risk of drug-drug interactions, and lower pill burden.
We will evaluate the efficacy of switching patients who are virologically suppressed on a second-line regimen of PI/r plus NRTIs to DTG plus NRTIs. This manuscript describes the study design, methods and baseline results after successfully completing study enrollment.
The primary objective of the Second-line Switch to DTG (2SD) study is to evaluate the non-inferiority of switching to a DTG containing regimen relative to maintaining a PI/r containing regimen among treatment-experienced, INSTI-naïve, virologically suppressed HIV-1 positive adults (≥18 years old) as determined by the proportion of participants having HIV-1 RNA ≥ 50 copies/ml at 48 weeks after enrollment.
Kenya has the fourth largest burden of HIV globally with close to 1.2 million people living with HIV,9 the majority of whom receive HIV treatment from public outpatient clinics. The Kenya National ARV Guidelines do not recommend performing DRT for patients failing an NNRTI-based first line regimen before switching to a PI/r-based second-line regimen,3 so resistance profiles are not available for most patients currently on PI/r-based ART.
The 2SD study takes place at four public hospitals in Kenya: Kenyatta National Hospital (KNH), Thika Level 5 Hospital, Kiambu Referral Hospital, and Jaramogi Oginga Odinga Teaching and Referral Hospital (JOOTRH). The four study sites currently provide ART to approximately 1,500 virally suppressed adults on PI/r-based second-line regimens.
2SD is an open-label, randomized, active-controlled, multi-centre non-inferiority trial. The trial enrolled participants from among people receiving outpatient HIV care at the four study sites in Kenya. Inclusion criteria were HIV-1 positive adults (≥18 years); on a second-line regimen of PI/r plus two NRTIs for at least 24 weeks; a treatment history of failing a prior NNRTI-containing ART regimen; and virally suppressed (plasma HIV-1 RNA < 50 copies/mL) for at least 12 weeks prior to enrollment. Women of childbearing potential (WOCBP) were not pregnant at the time of screening or enrollment and agreed to use effective contraception throughout the study period. Exclusion criteria included: any prior INSTI exposure; current use of disallowed concomitant therapy; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) at least five times the upper limit of normal in conjunction with hepatitis B virus (HBV) or hepatitis C virus infection; an estimated glomerular filtration rate of 50 ml/min or below in conjunction with HBV infection; advanced renal disease requiring dialysis; currently pregnant or breastfeeding; documented opportunistic infection within four weeks prior to enrollment; investigator opinion that the patient should switch from PI/r to DTG immediately for clinical reasons; or any condition or laboratory result which in the investigator’s opinion would interfere with assessments or completion of the study.
Based on a recommendation from the first Data and Safety Monitoring Board (DSMB) approximately four months after enrollment began and after 67% of participants were enrolled, the study protocol was amended to exclude new participants with baseline grade 3 or 4 lipid abnormalities as this would warrant a switch off of their PI/r immediately.
Screening investigations included HIV-1 RNA viral load, serum creatinine, ALT, AST, hepatitis B surface antigen (HBsAg) and urine pregnancy test for WOCBP. After the DSMB recommended the exclusion of patients with grade 3 or 4 lipid abnormalities, fasting lipids and fasting serum glucose were conducted as screening rather than baseline investigations.
Eligible participants were enrolled into the study within 28 days from their screening visit.
Participants were randomized in a 1:1 ratio using a computer-generated randomization sequence to either continue their pre-enrollment PI/r-based regimen or switch from PI/r to DTG. The study coordinator was blinded to participant details at the time of randomization; he was only provided with the participant identifier after all enrollment criteria were met and then generated the randomization number and study arm allocation. The randomization number and study arm allocation were provided to the site investigators who enrolled the participants. This is an open label trial and neither investigators nor participants were masked to treatment assignment.
Participants were randomized to continue their pre-enrollment PI/r (ATV/r, LPV/r or DRV/r) or switch from PI/r to DTG, while continuing the NRTIs from their pre-enrolment regimen. Changes to the NRTIs were allowed throughout the study period only for clinical indications following the Kenya National ARV Guidelines.3
For patients randomized to switch to DTG, the DTG was administered as a once-daily dosage without regard to meals, either as a single 50 mg tablet with a separate tablet of co-formulated AZT/3TC (300 mg/150 mg) one tablet twice daily, or as a single co-formulated tablet of DTG/3TC/TDF (50 mg/300 mg/300 mg) once daily, or as a single co-formulated tablet of DTG/3TC/ABC (50 mg/300 mg/600 mg) once daily. For patients randomized to remain on their pre-enrollment PI/r, the PI/r was administered as co-formulated LPV/r (200 mg/50 mg) two capsules twice daily, or co-formulated ATV/r (300 mg/100 mg) one tablet once daily. DRV/r is generally reserved for patients with prior PI/r failure in Kenya and no study participants were on DRV/r at enrollment. PI/r were recommended to be taken with food. For patients on PI/r, the NRTIs were administered as additional tablets of co-formulated AZT/3TC (300 mg/150 mg) one tablet twice daily, or co-formulated TDF/3TC (300 mg/300 mg) one tablet once daily, or ABC/3TC (600 mg/300 mg) one tablet once daily.
Study visits were scheduled at enrollment and weeks 4, 12, 24, 36 and 48. Visits were allowed to take place within ± 7 days from the specified follow-up date. Plasma HIV-1 RNA was measured at screening and weeks 4, 12, 24 and 48. Other investigations at screening, enrollment or follow-up included CD4 count, fasting lipid profile (total cholesterol, LDL and HDL cholesterol, ratio of total cholesterol to HDL cholesterol, and triglycerides), fasting serum glucose, serum creatinine, AST, ALT, HBsAg and complete blood count. For patients with protocol defined virological failure (PDVF), consisting of two consecutive HIV-1 RNA viral load results of 50 copies/ml or greater separated by at least two weeks, if their confirmatory viral load was 500 copies/ml or greater then genotypic drug resistance testing of integrase, protease and reverse transcriptase was attempted. All laboratory investigations were performed at accredited laboratories, at either the University of Nairobi Molecular and Infectious Diseases Research Laboratory or Lancet Laboratories. Point-of-care urine pregnancy testing was performed at all study visits for WOCBP.
Clinical screening for tuberculosis (TB) was performed at every visit following the Kenya National ARV Guidelines.3 Adverse events were graded using standard criteria.10 The HIV Treatment Satisfaction Questionnaire Status version (HIVTSQs) was administered at enrollment and week 48, and the HIVTSQ Change version (HIVTSQc) was administered at week 24. Treatment adherence was evaluated using pill counts of unused study drug at weeks 4, 12, 24, 36 and 48.
Study treatment was discontinued if requested by the participant, substitution of the DTG or PI/r was required due to drug-drug interactions or unacceptable toxicity, participant failure to comply with protocol requirements, pregnancy, or if deemed in the best interested of the participant for clinical reasons.
The primary outcome is the proportion of participants with plasma HIV-1 RNA of 50 copies/ml or greater at week 48 by the US Food and Drug Administration (FDA) snapshot algorithm. Secondary outcomes include proportion of participants with plasma HIV-1 RNA of 50 copies/ml or greater at week 24, proportion of participants able to maintain virological suppression with HIV RNA less than 50 copies/ml at weeks 24 and 48, change in CD4 count from baseline to weeks 24 and 48, and patient satisfaction as measured by the HIVTSQ at weeks 24 and 48.
Safety outcomes include clinical and laboratory adverse events at weeks 24 and 48, change in fasting blood glucose from baseline to weeks 24 and 48, change in fasting lipid parameters from baseline to weeks 24 and 48, and genotypic resistance to the study drug.
The sample size was calculated based on the primary endpoint of HIV-1 RNA of 50 copies/ml or greater at week 48 using the FDA snapshot method for the Intention-to-Treat Exposed (ITT-E) population. The ITT-E population is defined as all randomized participants who receive at least one dose of study drug, analyzed according to their randomization assignment. The sample size calculation assumed that the true difference in efficacy between treatment arms would be zero and that the overall virological failure rate would be 3% at week 48. A total of 766 participants (383 participants per study arm) were required to provide at least 90% power to demonstrate non-inferiority for the DTG arm, compared to the control arm, with a one-sided significance level of 2.5% and non-inferiority margin of 4%. The sample size was increased to 794 after the DSMB recommended excluding new participants with baseline grade 3 or 4 lipid abnormalities, to allow for fully powered sensitivity analysis if we excluded those who were already enrolled with grade 3 or 4 lipid abnormalities by the time of the DSMB recommendation.
For presentation of baseline results in this manuscript, participant demographic data and baseline characteristics using the ITT-E population were summarized by treatment group using descriptive statistics. Baseline continuous variables were summarized using medians (interquartile ranges), while categorical variables were summarized using counts and percentages. Ranksum tests were used for bivariate analysis between the DTG and PI/r groups on continuous variables while Chi-square or Fisher’s exact test as suitable were used for categorical variables, reporting the p values. All statistical tests were evaluated for significance at the 5% level.
Statistical methods planned for interim and final analysis include evaluation of efficacy, safety and patient satisfaction. The primary analysis will be performed after all enrolled participants complete their week 48 study visit or prematurely discontinue the study drug. Analysis of the primary endpoint will be performed for the ITT-E population using the FDA snapshot method. Non-inferiority for the primary endpoint will be established if the upper bound of the 95% confidence interval for the difference in proportion of participants with plasma HIV-1 RNA of 50 copies/ml or greater between groups is less than 4% in the ITT-E population. The primary endpoint will also be analyzed with the per-protocol (PP) population.
A secondary analysis will be performed for treatment success, defined as having HIV-1 RNA less than 50 copies/ml at week 48 in the ITT-E and PP populations, using the FDA snapshot method. Non-response is defined as any of the following: PDVF, death from any cause, loss to follow-up, consent withdrawal, or permanent change or interruption of the randomized treatment. Non-inferiority for the treatment success analysis will be demonstrated if the lower bound of the 95% CI of the difference between the groups is greater than –10%.
Other planned analysis include change in median CD4 count, lipid parameters, fasting glucose, and anthropometric measurements from baseline to weeks 24 and 48.
Safety analysis will be performed. Any AEs, grade 3 and 4 AEs, treatment-related AEs (all grade), treatment-modifying AEs (all grade), deaths, SAEs, and AEs occurring in at least 5% of participants will be described and compared by group.
Stata version 15.1 and R version 3.6.4 will be used to perform calculations and analyses.
An international DSMB is monitoring the main safety and efficacy outcome measures and overall conduct of the trial, and an independent study monitor conducted site initiation visits and will continue to monitor the study to conclusion.
The study was approved by the joint ERC of KNH and University of Nairobi and by the ERC of JOOTRH. All participants provided written informed consent. This clinical trial is registered at clinicaltrials.gov, NCT04229290.
Between February 10 and September 3, 2020, 1,114 adults were screened for eligibility and 795 were randomized, and 791 received treatment and are included in the ITT-E population: 397 switching to DTG and 394 to continue their pre-enrollment PI/r (Figure 1). Of the 319 that were screened and not enrolled, 195 did not meet eligibility criteria, 89 declined to participate, 33 did not return for randomization within 28 days from screening and two did not return for randomization before study recruitment was closed. There was no difference in gender or age distribution between the enrolled study participants and those who provided consent but were not enrolled (p = 0.57 and 0.84 respectively). Four randomized participants did not receive treatment: 2 withdrew consent (1 in each arm) and 2 relocated out of the study catchment area (both in the PI/r arm).
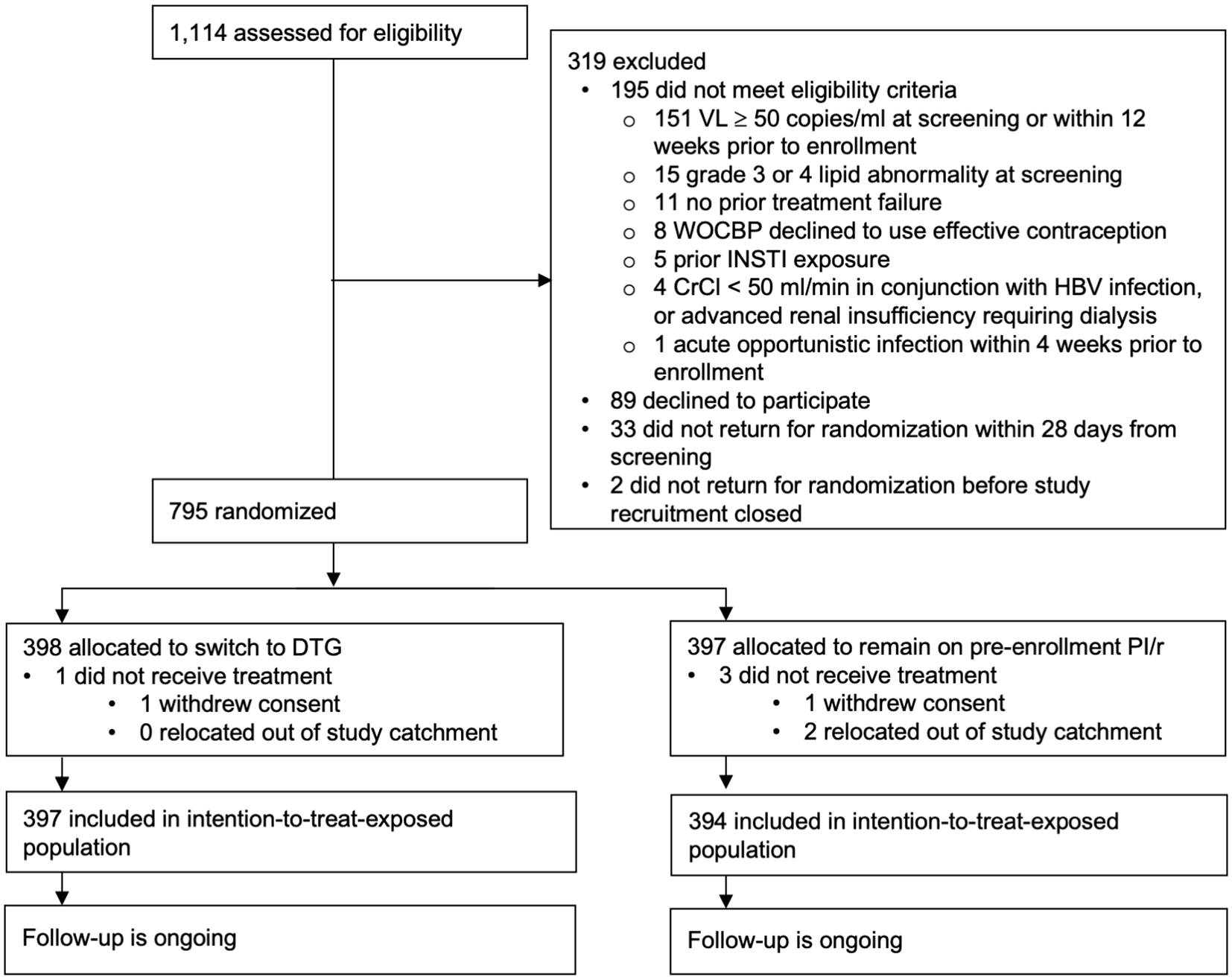
CrCl=creatinine clearance; DTG=dolutegravir; HBV=hepatitis B virus; INSTI=integrase inhibitor; PI/r=ritonavir-boosted protease inhibitor; VL=plasma HIV-1 RNA viral load; WOCBP=woman of childbearing potential.
Table 1 presents the baseline characteristics of the study participants. In summary, all participants were black, 524 (66.3%) were female, the median age was 46 years (range 19 to 74 years), and 25 (3.2%) were HBsAg positive. The median CD4 count at study enrollment was 423 cells/μl. Most participants (628 (79.4%)) were on ATV/r as their pre-enrollment PI/r, with the remainder on LPV/r. The median time on PI/r before enrollment was 5.4 years. The NRTIs at enrollment comprised TDF (52.8%), AZT (42.7%) and ABC (4.4%); all participants were also on 3TC. Baseline laboratory abnormalities of grade 2 or higher included 353 (44.6%) participants with reduced creatinine clearance (CrCl), 77 (9.7%) with dyslipidemia, 31 (3.9%) with impaired fasting glucose, and 43 (5.4%) with anemia. Baseline characteristics were balanced between treatment arms.
| DTG group (n=397) | PI/r group (n=394) | Total (n=791) | p-value | |
|---|---|---|---|---|
| Age (years)* | 46 (19-74) | 46 (19-72) | 46 (19-74) | 0.65 |
| Sex | ||||
| Women | 268 (67.5%) | 256 (65.0%) | 524 (66.3%) | |
| Men | 129 (32.5%) | 138 (35.0%) | 267 (33.8%) | 0.45 |
| Race: Black | 397 (100%) | 394 (100%) | 791 (100%) | 0.99 |
| Body-mass index (kg/m2) | 25.2 (21.7-28.8) | 24.4 (21.2-28.5) | 24.8 (21.5-28.6) | 0.11 |
| HBV co-infection | 13 (3.3%) | 12 (3.0%) | 25 (3.2%) | 0.85 |
| CrCl (mL/min) | 94.6 (80.1-116.4) | 93.8 (74.0-121.1) | 94.5 (77.5-117.9) | 0.74 |
| Grade 2-4 CrCl abnormality | 174 (43.8%) | 179 (45.4%) | 353 (44.6%) | 0.65 |
| Fasting TC/HDL ratio | 3.0 (2.3-3.8) | 2.9 (2.2-3.7) | 2.9 (2.3-3.8) | 0.35 |
| Grade 2-4 lipid abnormality)** | 55 (13.9%) | 61 (15.5%) | 116 (14.7%) | 0.52 |
| Fasting blood glucose (mmol/L) | 4.7 (4.3-5.2) | 4.7 (4.2-5.2) | 4.7 (4.2-5.2) | 0.78 |
| Grade 2-4 fasting glucose abnormality | 19 (4.8%) | 12 (3.1%) | 31 (3.9%) | 0.21 |
| Hemoglobin (g/dL) | 13.8 (12.6-14.9) | 13.6 (12.3-14.9) | 13.7 (12.4-14.9) | 0.46 |
| Grade 2-4 hemoglobin abnormality | 22 (5.5%) | 21 (5.3%) | 43 (5.4%) | 0.90 |
| HIV-1 RNA < 50 copies/ml | 397 (100%) | 394 (100%) | 791 (100%) | 0.99 |
| Median CD4 count (cells/μL) | 438 (311-603) | 397 (293-566) | 423 (306-589) | 0.08 |
| Category CD4 count (cells/μL) | 0.31 | |||
| <50 | 0 (0%) | 1 (0.3%) | 1 (0.1%) | |
| 50-199 | 28 (7.1%) | 34 (8.6%) | 62 (7.8%) | |
| 200-349 | 102 (25.7%) | 120 (30.5%) | 222 (28.1%) | |
| 350-499 | 114 (28.7%) | 107 (27.2%) | 221 (27.9%) | |
| >500 | 153 (38.5%) | 132 (33.5%) | 285 (36.0%) | |
| Time on PI/r regimen (years) | 5.5 (3.4-7.6) | 5.4 (3.1-7.8) | 5.4 (3.2-7.7) | 0.72 |
| Baseline PI/r | 0.15 | |||
| ATV/r | 307 (77.3%) | 321 (81.5%) | 628 (79.4%) | |
| LPV/r | 90 (22.7%) | 73 (18.5%) | 163 (20.6%) | |
| Baseline NRTI | 0.69 | |||
| TDF/3TC | 206 (51.9%) | 212 (53.8%) | 418 (52.8%) | |
| AZT/3TC | 175 (44.1%) | 163 (41.4%) | 338 (42.7%) | |
| ABC/3TC | 16 (4.0%) | 19 (4.8%) | 35 (4.4%) |
The 2SD study will provide important information on the efficacy of switching from a PI/r to DTG among virally suppressed, treatment experienced, INSTI naive, HIV-1 positive adults without prior information on resistance to NRTIs in the regimen. DTG has been found to be non-inferior or superior to boosted PI both for people who are treatment naïve11–13 and experienced,5,6 including from among a population with extensive NRTI resistance.6 Switch strategies from a boosted PI or NNRTI to DTG among virally suppressed adults without known or expected NRTI resistance have been found to be efficacious and safe.7,8 However, the 2SD study is the first randomized trial evaluating a switch strategy from PI/r to DTG for virally suppressed adults who have previously failed NNRTI-based regimens and with unknown NRTI resistance. If this strategy proves safe and efficacious, it will provide an opportunity to optimize ART regimens for approximately 75% of adults currently on PI/r-based regimens in Kenya and inform other large HIV programs in sub-Saharan Africa and globally.
The most prominent laboratory abnormality at study enrollment was the high proportion of participants (44.6%) with grade 2 or greater decrease in renal function. The prevalence of renal dysfunction among other HIV-positive cohorts on ART in sub-Saharan Africa is consistently high.14–17 The use of ATV/r and LPV/r have both been associated with increased risk of chronic kidney disease in large observational studies18,19 and TDF-associated renal toxicity also occurs more often when TDF is co-administered with a boosted PI,20 so our study population may have been at even higher risk for renal dysfunction having been on PI/r-based ART regimens for a median of 5.4 years before enrollment and with half of them also on TDF.
We also noted that 9.7% of participants had grade 2 or greater lipid abnormalities and 3.9% had a grade 2 or greater abnormality in fasting glucose. The Kenya National ARV Guidelines recommend baseline and annual testing of lipid profiles and glucose for all patients on ART,3 however these metabolic derangements were previously undetected before study enrollment for all participants. This may represent a challenge with comprehensive monitoring of patients on ART in low-income countries, and raises the potential merits of targeted investigations for populations at increased risk.
A strength of this study is that the majority participants are female, which is representative of the population receiving HIV treatment in Kenya and globally. Another strength is the relatively open eligibility criteria which increases the generalizability of study findings. We enrolled 71% of the patients screened, and of those who did not meet eligibility criteria, 77% were because they were not virally suppressed at the time of screening. So most virally suppressed adults on PI/r-based second-line therapy in Kenya would likely meet study eligibility criteria.
The primary limitation of this study is the open-label design. Participants are aware of whether they are continuing a regimen to which they have already become accustomed, or switching to a new regimen, and this could influence how participants attribute new symptoms during the study.
Harvard Dataverse. Replication Data for: Switching Treatment-Experienced, Integrase Inhibitor-Naïve, Virally Suppressed HIV-1 Infected Adults from Ritonavir boosted Protease Inhibitors to Dolutegravir: An Open-Label Randomized Controlled Trial. DOI: https://doi.org/10.7910/DVN/22RYAD.21
This project contains the following underlying data:
- This is the baseline dataset from an open-label, randomized, active-controlled, multi-centre non-inferiority trial conducted by University of Nairobi. The trial enrolled 791 participants ≥ 18 years old from among people receiving outpatient HIV care at the four study sites in Kenya. The variables contained in this dataset are; demographics, ARV history, body mass index, and baseline laboratory investigations (viral load, CD4, blood sugar, hemogram, HBV co-infection, renal function and lipids).
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Harvard Dataverse. CONSORT checklist. DOI: https://doi.org/10.7910/DVN/22RYAD.
The CONSORT flowchart is presented as Figure 1 in this manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)