Keywords
Dysbiosis, gut microbiota, metabolites, gut-brain axis, inflammation
This article is included in the Global Public Health gateway.
Dysbiosis, gut microbiota, metabolites, gut-brain axis, inflammation
Dysbiosis leads to structural and functional changes in the gut microbiome. If dysbiosis contributes to chronic inflammation by increased abundance of pathogenic flora it is termed as a gain of function dysbiosis. Loss of beneficial microbes promotes the development of chronic diseases such as irritable bowel disease (IBD), obesity, and others; this is known as loss of function dysbiosis. Moreover, increased abundance of pathogenic flora and decreased abundance of beneficial flora can occur simultaneously and lead to the onset of various diseases.1 The altered profile of gut microbiota is correlated with intestinal diseases such as coeliac diseases, IBD, and extraintestinal diseases including asthma, allergy, cardiovascular disease, chronic kidney disease, obesity, colorectal cancer, and metabolic syndrome (Figure 1). Dysbiosis might interfere with the gut-brain cross-talk, leading to the development of mental illnesses and neuro-disorders, including Alzheimer’s disease, Parkinson’s disease, anxiety, and depression.1 Commensal bacteria may affect brain functioning through γ-aminobutyric acid (GABA), directly influencing both immune and neural receptors within the enteric nervous system (ENS) and central nervous system (CNS). Alterations in central GABA receptor expression are implicated in the pathogenesis of anxiety and depression.2,3 Additionally, recent studies confirm that gut microbiota dysbiosis may contribute to mood swing disorders as well.4 Dysbiosis in neonates is a predisposition to energy and immune imbalance and metabolic syndrome.5 Dysbiosis also leads to increased production of pro-inflammatory cytokines. Inflammation-induced microenvironmental changes drive oxidative stress, generation of ROS and DNA damage, thereby leading to mutation and cancer. Gut microbiota imbalance, therefore, seems to be a root cause of chronic illnesses. However, understanding how gut microbial modulation causes various diseases is still in its infancy. This review is an effort to holistically review multiple aspects of dysbiosis and its role in disease initiation and progression.
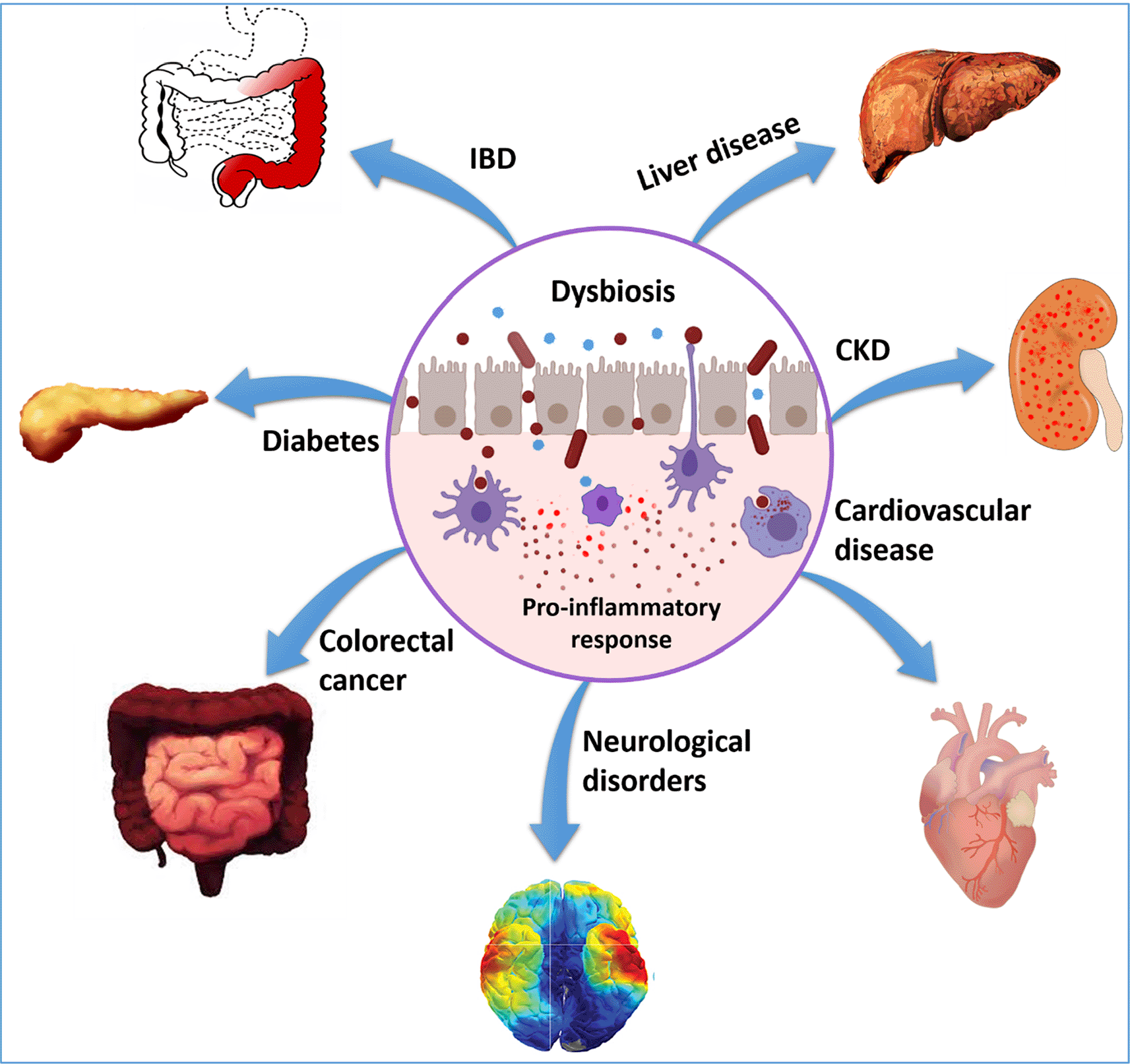
Dysbiosis is the change in the composition of the microflora of the gut leading to an increase in the abundance of pathogenic bacteria and a decrease in the abundance of beneficial micro-organisms. Therefore, the loss of commensals, bloom of pathobionts, and loss of diversity are characteristics of dysbiosis.6 Dysbiosis leads to changes in taxonomical composition as well as the metagenomic functions of the microbial communities. For example, the transformation of short-chain fatty acids (SCFA)-producing microbiota to toxic metabolite-producing microbiota disrupts microbial homeostasis and hampers the crosstalk between gut and brain, eventually leading to the development of various chronic diseases.
Gut microbiota is composed of various functional groups of microorganisms that conduct metabolic functions. These groups contain SCFA-producing bacteria (acrogens), sulfate-producing bacteria (methanogens), lactate-utilizing and producing bacteria, vitamin-producing bacteria and amino acid-, bile acid-using bacteria.7 The quantitative and/or qualitative change in the composition of these groups is known as taxonomic dysbiosis, whereas a change in metabolic profile is known as functional dysbiosis. Taxonomic dysbiosis has previously been studied through conventional microbiological techniques and recently using metagenomics.8,9 Mass spectrometry methods are used to study functional metabolomics. Both aspects of dysbiosis are discussed briefly in the following sections.
Taxonomic dysbiosis has been correlated with many diseases from common illnesses such as abdominal pain, bloating, and diarrhea, to chronic diseases such as IBD, CRC, diabetes, heart ailments, HIV, liver diseases, Alzheimer’s disease, and many more. Antibiotics that create an unfavorable environment for anaerobic gut flora provide a niche that is utilized by Clostridium difficile. C. difficile secretes toxins, which causes bloating and pseudomembranous colitis.10 Decreased abundance of members of the Streptococcus genus is associated with diarrhea.11 On the other hand, a reduction in protective bacterial abundance (Ruminococcaceae, Lachnospiraceae, Bifidobacteria, Firmicutes,and especially Faecailbacterium prausnitzii) and increased abundance of Enterobacteriaceae including E. coli, Pasteurellacaea, Veillonellaceae, and Fusobacteriaceae have been strongly correlated to IBD.12–14 Dysbiosis affects the functioning of the liver as well. In patients having non-alcoholic fatty liver disease, decreased abundance of Faecalibacterium prausnitzii and increased abundance of Proteobacteria, Enterobacteriaceae, and E. coli are reported.15 In contrast, chronic alcohol consumption can cause leaky gut and decreased gut bacterial diversity, which might be the leading cause of alcoholic liver disease.16 The overabundance of Chlamydophila pneumoniae, Helicobacter pylori, and Porphyromonas gingivalis is correlated with increased incidence of cardiovascular disease, although Bacteroides, Lactobacillales, and Clostridium are reported as potential markers of coronary heart disease.17,18 Intestinal epithelial cells might have extensive effects in terms of disease development and/or progression due to constant exposure to gut microbiota. Hence, the roles of gut microbiota and dysbiosis are precisely studied in CRC. Various studies have reported that Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis (ETBF), B2 subtype of E. coli, Salmonella, and Enterococcus faecalis could facilitate the development of CRC.19–24 Gut microbiota also plays a vital role in developing neurodevelopmental disorders, broadly known as autism spectrum disorders (ASD). Autistic children have distinct microbiota with less diversity. There is a significantly decreased abundance of Prevotella, Coprococcus, and unclassified Veillonellaceae genus and increased abundance of Bacteroides vulgatus and Desulfovibrio species in fecal samples.25 Gut bacterial biomarkers in various diseases are summarized in Table 1. Emerging studies have linked gut microbiota dysbiosis with many other diseases such as arthritis, bone disorders, diabetes, bad sleep. However, further studies are needed to elucidate microbial signatures and mechanisms of action.
Gut bacterial biomarkers in various diseases.
| Disease | Bacteria | Abundance in the patient compared to healthy control | No. of subjects/samples | Sequencing technology | Reference |
|---|---|---|---|---|---|
| Gestational diabetes mellitus | Enterobacteriaceae, Enterococcaceae Lachnospiraceae | Lower Lower | 48 HC, 59 GDM patients | 16S rRNA gene sequencing | (26) |
| Crohn’s disease | Actinomyces, Bifidobacterium Bifidobacterium adolescentis, Dialisterinvisus, Collinsellaaerofaciensand butyrate producing bacteriasuch as Blautiafaecis, Roseburiainulinivorans, Ruminococcus torques, Clostridium lavalense, Bacteroides uniformis and Faecalibacterium prausnitzii | Higher Lower | 10 HC, 10 inactive CD patient 55 HC, 68 CD patient | 16S rRNA gene sequencing Denaturing gradient gel electrophoresis (DGGE) | (27,28) |
| Colorectal cancer | Enterotoxigenic Bacteroides fragilis (ETBF), Fusobacterium nucleatum, Parvimonas micra and Campylobacter jejuni Bifidobacterium longum | Higher Lower | The cohort of 44 individuals | Whole-genome shotgun metagenome sequencing | (29) |
| Autism spectrum disorders | Firmicutes Alistipes, Bilophila, Dialister, Parabacteroides, and Veillonella, Collinsella, Corynebacterium, Dorea, and Lactobacillus | Higher | 40HC, 40 autistic patients | Pyrosequencing | (30) |
| Dementia | Bacteroides (enterotype I) Enterotype III | Higher Higher | NA | T-RFLP based 16S rRNA gene sequencing | (31) |
| Parkinson’s disease | Butyrivibrio, Pseudobutyrivibrio, Coprococcus, and Blautia Veillonella | Lower Higher | 51HC 64 PD patients | 16S rRNA gene sequencing | (32) |
| Necrotizing enterocolitis | Uropathogenic E. coli, Klebsiella spp., Enterobacter cloacae | Higher | 144 preterm, 22 term infants | 144 preterm, 22 term infants | (33) |
Metabolically active microbiota contributes to health by biotransformation of polymers into compounds that the host can utilize.34 Disturbance in microbial metabolism known as metabolic dysbiosis is the root cause of many diseases. Metabolic dysbiosis is characterized by metabolic abnormalities in the blood, urine, or feces.7,35 As metabolic dysbiosis is not necessarily followed by taxonomic dysbiosis, a completely different approach known as metabolomics is utilized to assess metabolic profiles.36 A recent study showed a clear separation between dysbiotic and non-dysbiotic IBD patients in terms of metabolic profiles. The dysbiotic IBD microbiome secretes about 122 distinct metabolites that are statistically different from non-dysbiotic microbiomes.37 Dysbiosis can have pro-atherosclerotic effects by modifying the production of a plethora of atherogenic metabolites, in addition to the metabolism-independent pathway.38 Dysbiosis has been demonstrated to impact bile acid (BA) metabolism as well as the synthesis of butyrate and trimethylamine-N-oxide (TMAO).39–41 Therefore, metabolites act as bacteria-derived signals in driving cardiovascular diseases.42 Dysbiosis can worsen systemic inflammation, which plays an essential role in chronic kidney disease (CKD).43 An increase in the abundance of uricase, urease, p-cresol, and indole-producing bacteria plays a crucial role in the initiation and progression of kidney failure.44 Larraufie et al. showed that gut bacteria-mediated production of peptide-YY (PYY) is important in appetite and energy expenditure.45 Dysbiosis can lead to the altered secretion of peptide YY (PYY) which promotes hypertension and obesity, both of which are the most common contributors to CKD.46 Another study conducted by Bajaj et al. reported a distinct metabolic profile of chronic alcoholic cirrhotic patients compared to non-alcoholic cirrhotic patients. Active drinkers have several significantly reduced amino acids such as ornithine, threonine, serine, and high-energy molecules such as citrate, malate, phosphate.47 An increase in the production of toxic secondary bile acids has been reported in dysbiosis-mediated liver disease.48 Nugent et al. reported an increase in prostaglandin E2, an inflammatory metabolite, and a decrease in diketogulonic acid, 5-oxoproline, antioxidant metabolites in adenoma cases than non-adenoma cases. The study determined 23 metabolites that distinguish adenoma cases from the controls.49 Various other studies have reported a high level of TMAO in body fluids of CRC patients.50 Metabolomics that provides metabolic profiles reflect the functional activity of the microbiota.51 Therefore, metabolomics can provide helpful metabolite biomarkers for the identification of diseases at an early stage (Table 2).
Gut bacterial metabolite markers in different diseases.
| Disease | Metabolites | Correlation with disease risk/severity | Species | Sample | Technique | Reference |
|---|---|---|---|---|---|---|
| Gestational diabetes mellitus | Trehalose and 3- dehydroshikimic acid 5-Hydroxyindoleacetic acid and valine | Positive correlation Positive correlation | Human | Urine | GC/MS | (26) |
| Positive correlation | Creatine, 1-palmitoleoylglycerol, urate, 2-hydroxybutyrate/2- hydroxyisobutyrate, xanthine, xanthurenate, kynurenate, 3-(4- hydroxyphenyl) lactate, 1- oleoylglycerol, 1- myristoylglycerol, dimethylglycine, and 2- hydroxyhippurate (salicylurate) | Positive correlation | Human | Plasma | (52) | |
| Cardiovascular disease | TMAO, Phenylacetylglutamine | Positive correlation | Human | Plasma, serum | UHPLC-MS/MS | (53) |
| Kidney disease | TMAO, Indoxyl sulfate, p-cresol sulfate | Positive correlation | Human | Plasma | LC/MS | (54,55) |
| IBD | SCFA | Negative correlation | Human | Fecal | LC/MS | (56) |
| Colorectal cancer | TMAO, Deoxycholic acids, Hydrogen sulfide, N-nitroso compounds, Polyamines | Positive correlation | Human | Serum, plasma. urine | (34,39,57,58) | |
| Autism | Propionic acid, 2-ketoglutaramic acid Acetic acid, Butyrate | Positive correlation Negative correlation | Rats, children Human | Serum Feces | LC/MS, HPLC/UV detector | (59–61) |
| NAFLD | Betaine, Betaine/choline ratio TMAO | Negative correlation Positive correlation | Human | Serum | HPLC-MS/MS | (62) |
| Alzheimer’s disease | TMAO | Positive correlation | Human | CSF | UHPLC-MS | (63) |
| Parkinson’s disease | Glutamic acid, Vitamin B3 and B5 Cadaverine, phenylalanine | Negative correlation Positive correlation | Human | Feces | GC-MS | (64) |
Controlling the interaction with the microbiota accounts for a significant portion of the intrinsic function of the immune system. As a result, gastrointestinal (GI) tract colonized by the most beneficial microorganisms serves as the interface with the highest number of immune cells. In response, we see a dominant effect of the healthy microbiota on the immune system that further reinforce barrier immunity and hence their containment to defend their ecological niche.65 Minimizing contact between microorganisms and the epithelial cell surface, decreasing tissue inflammation, and microbial translocation is a key approach the host uses to preserve its homeostatic connection with the microbiota. The combined activity of mucus, epithelial cells, immunoglobulin A (IgA), immune cells, and antimicrobial peptides in the gastrointestinal system, which has the highest density of commensals, achieves this segregation.66 Pattern recognition receptors (PRR) regulate microbial colonization.67 Dysbiosis affects levels of zonula occludens toxin, which in turn increases the intestinal permeability by disengagement of a tight junction protein complex which increases the entry of bacteria or bacterial components into lamina propria.68 This compromised intestinal barrier paves the way for bacterial cell wall parts such as lipopolysaccharide (LPS) endotoxin or whole microbes to enter the systemic circulation, which triggers the release of inflammatory cytokines from immune cells through T-cell receptor (TLR) activation. Humans can develop an inflammatory, immunological response to even minor increases in blood LPS levels (0.5–1.0 ng/kg).69 Thereby, dysbiosis leads to chronic, low-grade inflammation. Dysbiosis-induced gut permeability and inflammation are risk factors in various diseases such as IBD, advanced liver disease, CKD, diabetes, and CRC. On the other hand, through bacteria-derived metabolites, dysbiotic microbiota hijacks the host’s immune system and alters inflammasome signaling, modulates TLR signaling, and degrades secretory IgA (sIgA).70 In addition, some bacterial species such as segmented filamentous bacteria (SFB), Bacteroides fragilis, Clostridia spp, and Mucispirillum spp. directly control the development and differentiation of the immune system. SFB induces IgA production and activates Th17 cells through interleukin (IL)-23, IL-22, and serum amyloid A protein by direct attachment to the intestinal epithelium.71,72 On the other hand, most other bacteria use SCFAs, tryptophan metabolites, as communication mediators between microbiota and immune cells. These metabolites regulate regulatory T cells (Treg) homeostasis. A decrease in the abundance of SCFA-producing bacteria in dysbiosis creates imbalanced Treg production and inflamed mucosa.73 B. fragilis regulates the development of Treg cells through polysaccharide A.74
The human GI tract is essentially sterile in utero, so exposure to microbes during delivery and in the environment immediately following birth is key to the establishment of the microbiota. Colonization in the intestine starts right after birth, which depends on the mode of delivery and evolves as we grow. Cesarean section (C-section)-delivered infants are colonized with environmental and skin microbes (Staphylococcus, Streptococcus, Veillonella, Propionibacterium, E. coli, Salmonella, Klebsiella pneumoniae), whereas infants delivered through the vagina contain maternal vaginal and urogenital tract microbiota (Lactobacillus, Bacteroides, Prevotella, Bifidobacterium).75 Although this difference in microbial composition decreases with time, C-section birth is linked to a delay in the colonization of some taxa.76 The microbial abundance increases up to six-fold in the first few weeks after birth and continues to evolve in the first few years. The postnatal factors in microbiota development include consumption of antibiotics, breastfeeding, host genetics, and environment. The infant to adult microbiome transition starts when solid food is introduced into the diet, which stabilizes after approximately three years of life. Microbial communities that colonize in the initial stages shape our immune system and drive adaptive immunity development.77 Infant gut dysbiosis is characterized by a significant imbalance between beneficial and potentially pathogenic bacteria in the GI tracts of newborn babies. A recent study conducted a metagenomics survey and showed that 90% of US infants have signatures of dysbiosis, high levels of bacteria associated with gut inflammation, and antibiotic resistance genes. Overall, 93% of the microbiome belonged to 10 bacterial families and most of them are pathogenic bacteria (Enterobacteriaceae, Streptococcaceae, Clostridiaceae, and Staphylococcaceae).78 Pre-term infants have more microbiome instability, immature gut epithelium, and severe dysbiosis (higher proportion of Gammaproteobacterial than in-term infants).79 With the advancement in high-throughput sequencing technology, researchers have started studying the microbiome of the mother before birth. They proposed that colonization in mothers happens long before birth, with the mothers actively passing on to the offspring. Therefore, it is imperative to make the mother’s gut microbiome healthier with beneficial commensals rather than waiting until birth.80
Although the long-term effects of intestinal dysbiosis are unknown, some data suggest that it can have an immediate impact on an infant's health and contribute to disease vulnerability later in life. Infants with dysbiosis are vulnerable to various diseases such as necrotizing enterocolitis (NEC), sepsis, neurodevelopmental impairment, colic, atopic diseases, and diabetes. Studies proposed that neonatal gut dysbiosis may lead to the development of neonatal sepsis based on the hypothesis that early-colonized pathogenic bacteria in the gut may lead to an invasive infection.81 Pre-term infants with low birth weight and sepsis had lower microbial diversity than term infants and pre-term infants without sepsis. The pre-term infants without dysbiosis had chances of developing with increasing Proteobacteria and decreasing Firmicutes after a few weeks.82 Imbalance of gut flora in neonates and activation of the TLR4 pathway are also proposed as leading contributors of NEC, one of the most threatening GI diseases in newborns. Infants who suffer from NEC have lower microbial diversity and a higher abundance of Proteobacteria and Firmicutes.83 Breastfeeding promotes the development of a healthy microbiota in babies, which has long-term impacts on their health and wellbeing. Even if only carried out for a brief time, breastfeeding has been shown to improve intestinal dysbiosis in newborns.84 According to a study conducted in Europe, breastfeeding, mode of delivery, and country of birth shape the gut microbiota of infants at six weeks, where breastfed infants had predominant Bifidobacteria while formula-fed infants had a higher abundance of Bacteroides and Clostridium groups.85 Another study reported a significant increase in α diversity in fecal samples of breast-fed infants as compared to formula-fed infants.86 Although several studies confirmed the role of mode of delivery and breastfeeding in the development of infant gut microbiota, Cioffi et al. reported that breastfeeding has more lasting effects on the evolution of gut microbiota of children than mode of delivery.87 To untangle the relationship between breastfeeding, mode of delivery and gut microbiota evolution, more studies are needed with more samples, better coverage of the gut microbiota, and improved methodology.
Metabolic syndrome is a cluster of conditions that occur together, increasing one’s risk of heart disease, stroke, diabetes and other metabolic disorders. These conditions include increased blood pressure, elevated blood glucose level, excess body fat with centripetal obesity, and dyslipedemia. Studies on the association between antibiotic exposure in utero or during the perinatal period with childhood obesity and the triad of atopic dermatitis, asthma, and allergic rhinitis have been well reported.88 Metagenomic analysis in infants showed that under dysbiosis, fewer genes are available for the fermentation and biosynthesis of SCFAs, making them vulnerable to developing type 1 diabetes in postnatal life.89 Dysbiosis has been correlated with the development of respiratory allergies in neonates. Chua et al. confirmed that imbalanced gut microbiota in infants enriched in Ruminococcusgnavus developed asthma (airway inflammation). Asthma was characterized by increased Th2 cells, infiltration of eosinophils, and mast cells in colon and lung parenchyma.90 Similarly, infants suffering from acute dermatitis and obesity had lower bacterial diversity, suggesting the role of dysbiosis in disease progression.91,92
The gut microbiome and the brain communicate bidirectionally, each one potentially influencing the functioning of the other. This communication bridge is known as the gut-brain axis (GBA). The efficient functioning of the microbiota-gut-brain axis includes coordination among the CNS, autonomic nervous system (ANS) including sympathetic and parasympathetic branches, ENS, the immune system, metabolic signaling, and endocrine system93 (Figure 2). In vitro, in vivo, and human studies confirm a positive correlation between gut microbiome dysbiosis and functional changes in the brain.94 The microbiota can affect the CNS directly or indirectly through the modulation of neurotransmitters and neuroactive microbial metabolites such as short-chain fatty acids.
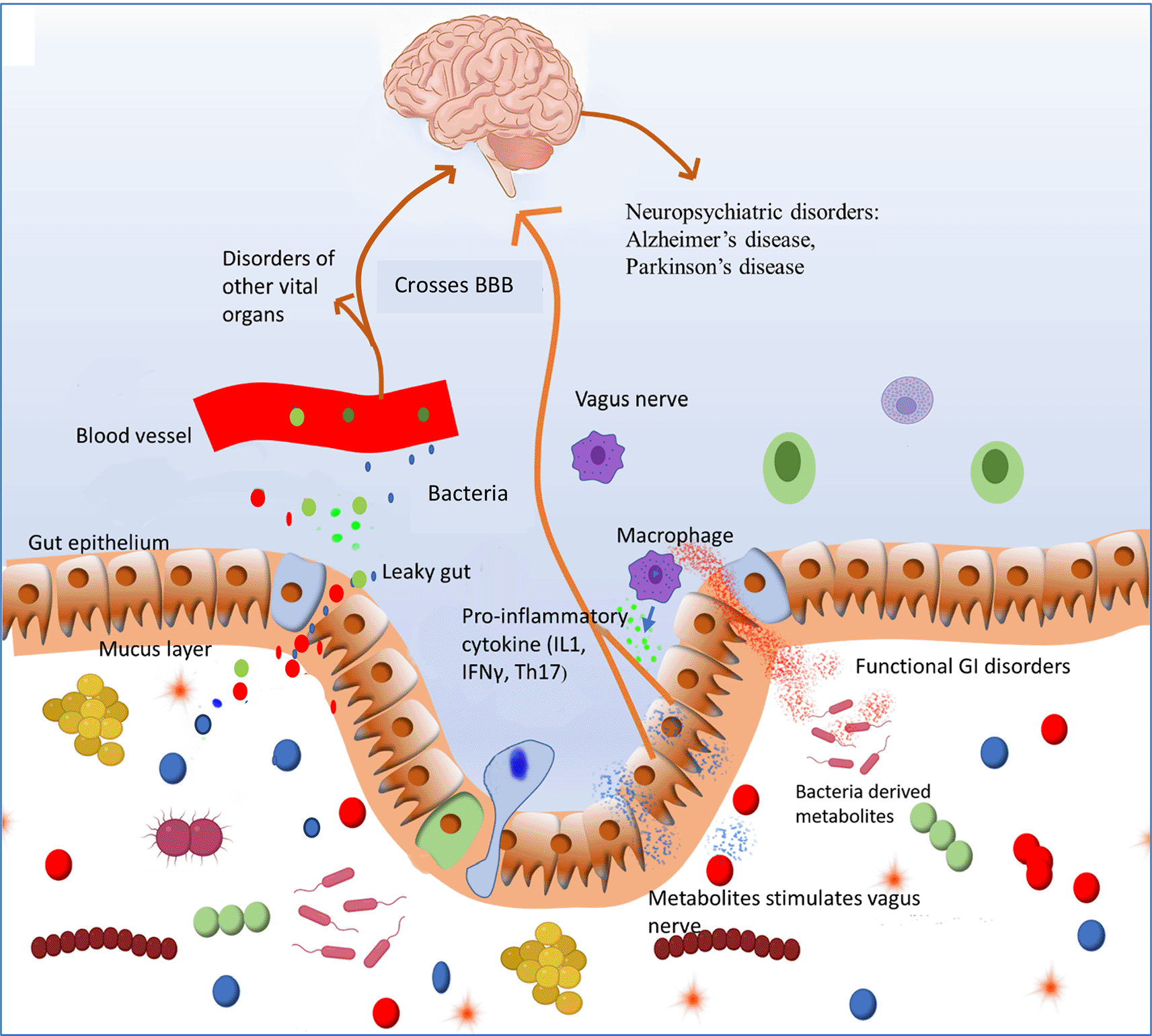
Pathobiont overgrowth induces inflammation and loss of barrier function that in turn promotesincreased translocation of bacterial components and even living bacteria into the host’s internal environment. This process will activate innateimmunity characterized by production of proinflammatory cytokines that define a state of systemic inflammation which contributes to functional GI disorders. Bacterial components such as metabolites enter systemic circulation and reach other organs where they can initiate the development of various chronic diseases. Bacterial metabolites can cross blood brain barrier (BBB) and modulate functioning of brain and could contribute to development of neurological disorders.
The vagus nerve sits at the epicenter of a crucial communication network, connecting the GI tract with the brain. Microbes interact with enteroendocrine cells to produce different classes of neurotransmitters (glutamate, acetylcholine, Gamma butyric acid, serotonin) and neuropeptides which activate the vagus nerve and send a signal to the brain.95 Although the exact mechanism by which the gut microbiome interacts with the vagus nerve is unknown, it is assumed that the vagus nerve can distinguish between harmful and non-pathogenic bacteria.96 The pro-inflammatory cytokine IL-1, produced in dysbiosis or sepsis, activates the vagus nerve. Vagal afferents, in turn, increase the release of glucocorticoids, which reduces peripheral inflammation. This shows that the vagus nerve has an anti-inflammatory response. Dysbiosis caused by chronic H. pylori infection causes TNFα to be upregulated, resulting in biochemical and behavioral disorders that persist even after the bacteria has been eradicated.97 Several CNS disorders, such as neuroinflammatory, neuropsychiatric, and neurodegenerative disorders, have severe inflammatory symptoms that are linked to gut microbiota dysbiosis.98 Therefore, the gut microbiota is involved in the regulation of CNS function, development, and host behavior.
Immune-related pathway
In addition to the protection of commensals and immunological acceptance of non-pathogenic microbes, the microbiota affects both innate and adaptive immunity. The absence of microbiota makes the host deprived of IL-22 which is crucial for host immunity to fight against enteric bacterial infections. The host defense system against pathogenic microbiota utilizes two pattern recognition receptor systems: toll-like receptors (TLRs) and nucleotide-binding oligomerization domain receptors (NODs). Both of these systems help macrophages to identify microbe-associated molecular patterns (MAMPs) expressed by gut microbes and produce pro-inflammatory cytokine IL-1β.67,99 IL-1β helps in the regulation of elimination of pathogens from the body. Mice with a broad-spectrum antibiotic-induced dysbiosis had decreased myeloid cell growth in the bone marrow, fewer circulating granulocytes, and were more prone to systemic bacteremia. According to this study, the gut microbiome increase production of immune cells to reduce bacterial infection.100 Another study showed a proinflammatory cytokine profile featuring elevated IFNγin rats having intestinal dysbiosis.101 Furthermore, gut microbiota maintains immunological homeostasis in the GI tract by harmonizing pro-inflammatory Th17 and anti-inflammatory Tregs, preventing intestinal and systemic inflammation.98 Dysbiosis can induce depressive-like behaviors, neuro-developmental disorders such as anxiety, autism, attention deficit hyperactivity disorder (ADHD), depression, and schizophrenia.102 Hence, gut microbiota maintains host health through various immune-related pathways and helps the host to get rid of any persisting bacterial infection.
Bacteria produce various small molecular by-products known as metabolites which help gut microbiota communication with the CNS. These metabolites activate the vagus nerve and send a signal to the brain which reciprocates through efferent nerves. GABA, the major inhibitory neurotransmitter in the brain, is produced by many Lactobacillus and Bifidobacterium species. Furthermore, Candida, Escherichia, and Enterococcus generate the neurotransmitter serotonin, whereas some Bacillus species synthesize dopamine.103 In the gastrointestinal tract, serotonin governs pain perception, motility, and secretion, whereas, in the brain, it links mood and cognition. Bacteria can also synthesize SCFAs, which stimulate the sympathetic nervous system and mucosal serotonin production, influencing the brain's memory and learning processes. They improve intestinal epithelial integrity, boost mucus production, modify gut motility, and have anti-inflammatory properties, such as nuclear factor κB inactivation and the stimulation of Treg cells.104–106
Neuroendocrine signaling (hormones)
Bacterial products such as SCFAs, tryptophan-derived metabolites, and peptidoglycans are known to stimulate enteroendocrine cells to produce several neuropeptides and hormones such as neuropeptide Y (NPY), peptide YY (PYY), glucagon-like peptide (GLP)-1, 2 cholecystokinins (CCK), substance P, α-melanocyte-stimulating hormone (αMSH), calcitonin-generated peptide (CGRP), vasoactive intestinal peptide (VIP) and adrenomedullin (AM).107,108 These hormones are proposed to have an important role in the regulation of the gut microbiota composition. These neuropeptides then enter the bloodstream to directly influence the ENS. The ENS sends signals to the CNS which in turn modulate intestinal functions via the hypothalamic-pituitary-adrenal (HPA) axis and via parasympathetic and sympathetic branches of the nervous system. On the other hand,in dysbiosis and stress conditions, the HPA axis is disturbed which stimulates enterochromaffin cells to release catecholamines, serotonin, cytokines to regulate gut functioning.103 Dysbiosis of gram-negative bacteria produces LPS, LPS-binding protein (LBP),and flagellin, which are potent endotoxins. LPS activates TLR4 and induces the production of pro-inflammatory cytokines such as TNF-α, IL-1, 6.109 Several neuropsychiatric disorders linked to gut dysbiosis and intestinal inflammation have been linked to altered amounts of (gut) neuropeptides, although it's unclear whether these neuropeptides are also affected in the GI tract. There is no evidence that these neuropeptides originate in the gut or that changed neuropeptide levels are linked to the progression of intestinal inflammation in these pathologies.
Despite the fact that several types of bacterial phyla dominate the human GI tract, such as Firmicutes, Actinobacteria, and Bacteroidetes, the overall composition of the human gut microbiota is highly variable, which gives rise to inter-individual variation in drug therapy responses.110 The effect of gut microbiota variations on pharmacodynamics and pharmacokinetics is known as pharmacomicrobiomics which deals with the interactions between the gut microbiome and xenobiotics.111 Gut microbiota affects drug efficacy by activating or inactivating pharmacological properties of drugs, or by producing drug-metabolizing enzymes involved in drug activation, inactivation or deconjugation.112 As a matter of fact, the gut microbiota can execute a variety of metabolic responses on medicines, drug metabolites, and other xenobiotics.113 Gut microbiota regulates drug-metabolizing genes. Claus et al. discovered that germ-free mice had lower levels of Cyp2c29, Cyp3a11, and Cyp8b1, necessary for drug metabolism. The expression level of these enzymes was normalized after 20 days of bacterial colonization.114
Dysbiosis has been linked to decreased efficacy of medicines along with increased chemotherapy toxicity.115 A paradox also emerges, in that chemotherapeutics may aggravate rather than alleviate any dysbiotic state, potentially resulting in serious drug toxicity.116 But there is a paucity of data on the effect of dysbiosis on drug efficacy. Some studies report that dysbiosis negatively affects the effectiveness of drugs used in chemotherapy. Yuan et al. showed that antibiotic-induced dysbiosis significantly reduced the efficacy of 5-Fluorouracil, a drug used for the treatment of CRC. Antibiotics reduced microbiota biodiversity and altered microbiota community. The pathogenic bacteria including Escherichia, Shigella, and Enterobacter significantly increased, while other commensal bacteria decreased.117 Cyclophosphamide is another cancer drug that stimulates antitumor immune responses. This drug induces the translocation of selected species of Gram-positive bacteria into secondary lymphoid organs. Such bacteria stimulate a subset of "pathogenic" Th17 (pTh17) cells to be produced, as well as memory Th1 immune response. Tumors were resistant to cyclophosphamide in antibiotic-treated animals without Gram-positive bacteria, and pTh17 responses were reduced.118 Therefore, the gut microbiota plays a crucial role in maintaining intestinal homeostasis, drug metabolism, and co-metabolism. However, depending on the microbiota composition, this impact may be beneficial or deleterious to the host.119
The contribution of the gut microbiota to drug metabolism and toxicity is astonishingly recognized but remains largely less studied due to the highly complex relationship between the gut flora and the host. Understanding the underlying mechanism of the gut microbiota in drug metabolism is necessary before any practical advances can be made in drug development or personalized medicine. Innovation in analytical methods is urgently needed to enhance the ability to recognize microbial functions, including metagenomics, metabolomics, and interdisciplinary knowledge integration. Studies of the effects of gut microbiota on drug metabolism and toxicity will provide new evidence for personalized medicine by manipulating the gut microbiota.
The field of microbiome research has evolved rapidly in the last decade and has become a topic of great scientific and public interest. Nonetheless, this extremely fast-growing discipline faces a variety of challenges such as data standardization, better coordination, and collaboration across the field of microbiome. Last but not least is finding the underlying mechanistic details.
In human studies, confounding factors like geographic location, race, diet, culture, health status,variation in the microbiome, and drug use lead to inconsistency in identifying microbiota associated with various diseases.27,28 Microbes are rarely annotated at the species or strain level using current sequencing and analytical methods. This leads to difficulty in the identification of disease-associated microorganisms because functional capacity differs amongst strains of the same species. The transcriptome and proteome are less studied in the majority of dysbiosis studies. The use of multi-omics technology in microbiome research could improve accuracy. Variation in the microbiome of animals that are otherwise genetically identical and incompatibility of gnotobiotic advanced technologies with omics technologies pose challenges in animal studies examining the dysbiotic microbiome. To validate predictions made in human studies and explore processes of host-microbiota interactions in dysbiosis-related metabolic disorders, the scientific community must overcome these technical difficulties and develop robust and standardized experimental systems.
The emergence of metagenome-wide association studies in the microbiome area over the last two decades has demonstrated that human biology is a holobiont biology. In other words, human biology is dependent on coexisting microbes, the majority of which live in the digestive system and generate bioactive chemicals, or activate host reactions that affect numerous physiological functions such as immunity, neurobiology, as well as metabolism.120 One popular theory proposes that the lack of diversity in our microbiome increases the risk of diseases.1 This study demonstrates that dysbiosis is a complex problem of the intestinal microbiota that is thought to play a role in the pathogenesis of IBD, as well as other diseases such as CRC and allergy disorders, although more research is needed to corroborate this theory. The involvement of numerous bacteria such as Fusobacterium nucleatum, ETBF, E. coli, and Klebsiella spp in the development of various health conditions has been reported using metagenomics of stool samples. In neonates, dysbiosis can lead to fatal infections such as NEC and developmental problems. Emerging novel biomarkers promise precise phylum and species discrimination, confirming evidence of microbial involvement in type 2 diabetes, obesity, metabolic disorders, IBD, and even cancer.121
Most research provides a snapshot of the microbiome at one or two time points, although, it is highly variable over time; therefore, more data on the short- and long-term patterns of the intestinal microbiome is needed. Most studies have utilized fecal samples for microbiota analysis since the collection of materials from the human intestine is difficult. The stool microbiota profile does not entirely reflect the gut microbiome as feces do not include all the microbes in the gut.122 As a result, non-invasive methods for collecting microbiota samples from various places along the intestinal system are required, as feces only represents a small portion of the gut microbiota. Metabolomics is used to investigate the metabolic characteristics of the functional microbiota. Dysbiosis causes metabolite concentrations to change, which can lead to crucial organ damage. TMAO is one such metabolite that has been linked to several diseases and is therefore a metabolite of concern. Researchers have used metabolomics to go beyond finding biomarkers to deciphering the mechanisms that underpin phenotypes.123
Future researchers must be aware of the multiple factors that influence the development of gut dysbiosis, including genetics, food, and environmental factors. Future researchers should be able to get closer to address dysbiosis and its related disorders. The ongoing effort of identifying the gut microbiota present in humans will provide a better understanding of the role of dysbiosis in health and disease.124
When it comes to the translational implications of microbiome research, it was recently discovered that, in addition to their well-known impacts on clinical variables, most medications influence the microbial, metatranscriptomic, and metabolomic profiles.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)