Keywords
A549, Diazaitidine, Mefenamic Acid, Molecular Docking, Synthesis
This article is included in the Cheminformatics gateway.
A549, Diazaitidine, Mefenamic Acid, Molecular Docking, Synthesis
Lung cancer is the leading cause of cancer-related death worldwide.1 Cancer treatment causes severe physical and mental exhaustion due to the high toxicity and ineffectiveness of commercially available anticancer drugs.2 Efficient targeting has led to the development of more potent and less toxic anticancer drugs in recent years.3
Cancer and inflammation are tightly connected, tumor development and growth are greatly influenced by inflammation. Nonsteroidal anti-inflammatory drugs (NSAIDs), among other anti-inflammatory medications, have been proven in several clinical investigations to affect the tumor microenvironment by preventing cell migration, promoting apoptosis, and increasing chemosensitivity.4 There has been a lot of focus on the role that inflammation plays in the onset and aggressiveness of several malignancies, including non-small cell lung cancer (NSCLC). Numerous studies have shown that NSAIDs help prevent cancer in animal models, in part because they can reduce the activity of the enzyme cyclooxygenase (COX). It has been demonstrated that cyclooxygenases, particularly COX-2, are important during various phases of carcinogenesis. Erlotinib, an EGFR tyrosine kinase inhibitor, was combined with a number of nonsteroidal anti-inflammatory medicines (NSAIDs) to create numerous novel anticancer medications.5 Nuclear transcription factors that have been shown to be overexpressed in cancer cells are inhibited by NSAIDs. Nuclear factor kappa B (NF-B) controls the expression of inflammatory and proliferative enzymes and proteins such as COX-2 and cyclin D1. Therefore, blocking the COX-2 enzyme may reduce the risk of cancer development, progression, and spread. Thus, inflammation can provide key mutations and a suitable environment for tumor growth.6
Due to their role in the treatment of inflammation and their cytotoxic activities, NSAIDs have become a popular target for drug design technology to create substances with improved COX-2 selectivity and fewer side effects.7
Mefenamic acid (MFA) is an anthranilic acid-class NSAIDs.8 In an era where other (NSAIDs) have proliferated, the clinical application of mefenamic acid has typically waned. It is a member of the fenamates and shares comparable modes of action and general toxicities with others (NSAIDs).9 However, mefenamic acid has distinct in vitro properties that may separate this medication from others. The basic sciences have created an opening for using mefenamic acid alone or in conjunction with other medicines for some novel indications. For instance, the effect of mefenamic acid on persistent, progressive brain disease needs additional exploration.10 Cellular mechanisms of action could be primarily utilized to influence cellular and then macroscopic transformation in various novel applications.11
Mefenamic acid has been demonstrated to be effective against human breast cancer (MCF-7), human bladder cancer (T24), human lung cancer (A-549), and other cancers by inducing apoptosis in human hepatoma cells (CHANG and HuH-7).12
A. Altai et al. successfully prepared two Ag(I) complexes containing mefenamic acid. Flow cytometry was used to study the apoptotic effects of complex and intracellular ROS production in MCF-7 cells. These XTT and LDH assays demonstrate that both complexes exhibit potent anti-proliferative activity with higher selectivity for cancer cells compared to normal cell selectivity.13
A major source of therapeutic medicines in medicinal chemistry is heterocyclic analogs based on nitrogen. Because biological targets and nitrogen easily form hydrogen bonds. Quaternary rings are becoming more crucial in drug development initiatives as bioactive scaffolds. They contribute to the advancement of physicochemical qualities as well as structural innovation to previously unknown regions of chemical space.14
Santos and co. completed an important step in the production of a series of 1,2-diazetidin-3-ones by the intramolecular N-H insertion of rhodium carotenoids. These substances have a mild growth-inhibitory effect on breast cancer cells.15
The strategy of this work was to incorporate this heterocyclic four-membered ring system into mefenamic acid to synthesize new derivatives and evaluate their anti-proliferative activity against the A549 cell line using the MTT method, with an emphasis on understanding how to bind to the EGFR and its possible effects on cancer cells, hoping to design new heterocyclic compounds that may help in the development of new potent anticancer agents with high safety.
FT-IR spectra were recorded with [Tensor 27 Brucker (USA), Shimadzu FT-IR KBr disc (Japan)], melting points were measured with glass capillaries on a Stuart melting point apparatus (UK), proton NMR (400.13, 300.81). MHz) and carbon NMR spectra (100.62, 75.65 MHz) were determined by Brucker (USA) using DMSO as solvent. Mefenamic acid and various aryl aldehydes from Hyperchem (China), SOCL2 from Thomas Baker (India) and phenylisocyanate from Sigma-Aldrich (Germany).
Mefenamic acid was dissolved in 15 ml of dry methanol at a ratio of 4.3 mmoles, and the mixture was chilled to a temperature between 0 and -5°C. To guarantee full solubilization, a small amount of tetrahydrofuran solvent was added. Thionyl chloride was gradually added to the solution dropwise until 0.32 ml (4.5 mmol) had been added. The mixture was heated in a water bath at 40°C for three hours, followed by eight hours of reflux stirring at 65°C. It was evaporated and then treated with 25 ml of ice water after it had warmed to room temperature. The product is recrystallized from 100% ethanol to produce a pure product, using a silica gel-coated TLC plate to monitor the reaction.16 The purity of the synthesized compounds was assessed through recrystallization and confirmed by determining a sharp melting point. Physical properties, Rf values, FT-IR, and NMR spectra interpretations of the synthesized compounds are illustrated in Table 1.
Compound [M2] was synthesized by dissolving 1 mmol of compound [M1] in 15 mL of absolute ethanol. Add an excess of 0.25 mL (5 mmol) of hydrazine hydrate, 99.99%, to the solution. The mixture was refluxed for 15 hours. After cooling, ice distilled water were added to precipitate the solution. Recrystallization from absolute ethanol gave compound [M2] a light yellow powder. Monitor the reaction with silica gel-coated TLC plates.17
Each aromatic aldehyde (1 mmol) was combined with 10 mL of 100% ethanol before adding 2-3 drops of glacial acetic acid to begin the synthesis of the compounds [M3a-e]. The mixture was then swirled for 30 minutes at room temperature. Then mix 1 mmol of compound [M2] with 15 ml of 100% ethanol for 30 minutes at room temperature, reflux the mixture for 3 to 6 hours at 75 °C. After filtering, washing with cold water, recrystallization from pure ethanol. Follow the development of the reaction on a TLC plate with silica coating.18
Anhydrous 1,4-dioxane (25 ml) was first mixed with 1 mmol of (M3a-e), and then 1.5 mmol of phenyl isocyanate was slowly added dropwise while maintaining a temperature between 0 and -5°C. The resultant mixture was refluxed for (5-7) hours after being agitated for 3 hours at room temperature. The resultant solid was washed with a solution of ethyl acetate: petroleum ether (1:1), filtered, and dried after the solvent had evaporated at room temperature. The process of recrystallizing was done from pure ethanol.14
The synthesis method of the compound M3a-e and M4a-e is shown in Figure 1.

GOLD Suite (v. 2021.2.0), a licensed CCDC tool, was utilized for the docking technique to examine the possible binding of certain 1,3-diazetidin-2-one derivatives to EGFR. A nonproprietary alternative that can be suggested are AutoDock and AutoDock Vina. Additionally, using the CCDC Hermes Visualizer program (version 2021.2.0), bond lengths, brief contacts, hydrogen bond interactions, and interactions between proteins and ligands were estimated. Nonproprietary alternative that can be suggested in this instance are PyMOL, jmol and ChimeraX. The ligands from Chem Sketch were transformed into SMILE names using the Swiss ADME program, and the physicochemical characteristics and pharmacokinetic statistics of molecular atoms were predicted. Additionally, using BOILED EGG,19 the lipophilicity and polarity of molecules are estimated.20 The component amino acids of the EGFR protein crystal structure now exist in ionized and tautomeric forms because hydrogen atoms have taken the role of water molecules in the structure. With default settings for all parameters, CheBio3D (version 16.0), MM2 force field, and CHEMPLP fitness algorithm were used to assess each proposed solution. Nonproprietary alternatives to CheBio3D are Avogadro, MarvinSketch, and BKChem. Additionally, the hydrogen atoms' dependence on distance and angle was taken into account when calculating the steric complementarity between the protein and its ligand. The candidate solutions are then evaluated, and the binding mode, docking position, and binding energy for each candidate solution are determined by a piecewise linear potential function. These findings assess how our created compounds interact with the EGFR's amino acid residues.21
Cell culture
A human lung cancer cell line (A549) from ATCC has been purchased by Al Mustansiriyah University Cell Bank Tissue Culture Research Center and is currently housed at the Faculty of Pharmacy.
Storage and resuscitation of cell line
For 24 hours, cells were kept frozen in liquid nitrogen (-80°C). Add 10 ml of fresh media after thawing at 37 °C, and the cells were separated via a centrifuge. The cells are then transfered to a 75 cm2 culture flask after resuspending them in 25 ml of fresh media.22
Cell maintenance
The cells are incubated at 37°C in 95% humidified air and 5% CO2, A549 cells were cultured in RPMI 1640 medium. The medium was supplemented with antibiotics such as penicillin-streptomycin-amphotericin B100X and 1% L-glutamine. An addition of 10% of fetal bovine serum (FBS) was made to enhance the medium. In sterile 75 cm2 flasks, the cells were grown and when 90% confluency was reached, they were subcultured by washing with 5 ml of phosphate-buffered saline (PBS) and detached from the bottom of the flask using trypsin solution that was incubated for 2 min at 37°C. Using a hemocytometer, check the cell count after transferring a cell suspension to a 50 ml conical tube and centrifuging at 1200 rpm for 3 minutes. After decanting the supernatant, resuspend cell pellets in supplemented growth medium. Complete growth medium should also be added in equal volume.23
Cell viability by MTT colorimetric assay
In an effort to uncover new information, we conducted an experiment to evaluate how synthetic chemicals [M4a-e] would impact the survival of lung cancer cell lines by administering the MTT colorimetric assay. Our approach involved dispensing 100 μl of a cell suspension, with a concentration of (5×103) cells/well, into each individual well of a 96-well flat-bottom tissue culture plate and leaving them undisturbed for a total of 24, 48, or 72 hours. After the first 24 hours, we introduced each chemical, with a 5 μm concentration, to the cells. Incubate the culture in a medium containing 10 μl of MTT solution (5 mg/ml MTT powder in PBS) at 37°C for two hours after the recovery period (24 h, 48 h, and 72 h). Cast aside the medium after 2 hours, and attribute 100 μl DMSO to each well, and put it in a dark room at room temperature for around 15-20 minutes. Using a Multiscan Reader, the optical density of each plate (well) was measured through a transmitted wavelength range of 560-600 nm. To calculate the cell growth inhibition (percent cytotoxicity), the formula [Percent inhibition = (A-B/A)*100] was implemented, with [A and B] representing the tested optical density substance and the standard (erlotinib), respectively. Performances were completed in duplicate.24
Determination of the half maximal inhibitory concentration (IC50)
Using the dose response curve, the IC50 of the test compound can be calculated. For in vitro MTT assays, IC50 is the concentration of the test chemical [M4a-e] required to reduce cell viability by 50%. Using the results of the in vitro MTT assay, IC50 values were calculated for the study drug 72 hours after exposure to the cells. The concentration ranges of the substances [N4a-c] used for the calculation of IC50 values were (500, 250, 125, 12.5, 62.5, 31.25, 15.62, 7.81, 3.90 and 1, 95 μm).
The identification of the synthesized compounds was made through the examination of their melting point and Rf values. The FT-IR analysis revealed that the hydroxyl group of mefenamic acid had disappeared, as seen from the absence of its broad peak at 2500-3200 cm-1. Instead, a signal was detected at 1732 cm-1 for compound (M1), indicating the presence of a carbonyl group of ester (-C=O) in place of the carboxylic acid (C=O) found in mefenamic acid. The stretching of the ester was observed at 1224 cm-1 for compound (M1), while the singlets of (-OCH3) of ester were detected at 3.87 (δ) in the 1H-NMR results. The broad peak of the hydroxyl group at 12.99 (δ) for mefenamic acid had disappeared. In the 13C-NMR results, a peak at 168.71 (δ) was observed for the carbonyl of the ester in (M1), while the methyl group showed a peak at 52.19 (δ).
Infrared analysis (FT-IR) of compound (M2) revealed the disappearance of a broad band at (1732 cm-1) associated with stretching of the carbonyl (C=O) of the (M1) ester and the appearance of a New bands are assigned to stretches of the amide carbonyl (C=O) and to the (-NHNH2) group of (M2) at (3351, 3329, 3188 cm-1). The 1H-NMR spectrum showed a broad singlet at 4.31 (δ) for the NH2 proton of the hydrazide and a singlet at 9.52 (δ) for the NH proton of the hydrazide (M2). 13C-NMR showed that the methyl ester peak disappeared and the amide carbonyl at (172.73) (δ) became visible.
The FT-IR characteristic absorption bands of (M3a-e) showed υNH stretching of amide at (3344-3325 cm-1) and combination band of υC=O stretching of amide and υC=N stretching at (1664-1622 cm-1). 1H-NMR spectra of compounds (M3a-e) showed singlet for N=CH-Ar (imine proton) at (8.34-8.88) (δ), and disappearance of the broad singlet for NH2 protons of hydrazide. 13C-NMR showed the peak of (-CH) imine at (~143-150) (δ).
The characteristic FT-IR absorption band of N-H stretching of secondary amides appears at (3325, 3286 cm-1) as two peaks due to hydrogen bonding; in the presence of hydrogen bonding, the N-H stretching splits into two peaks: one relatively low frequency peak and a higher frequency peak. Lower frequency peaks correspond to N-H stretching in hydrogen-bonded NH groups, while higher frequency peaks correspond to N-H stretching in non-hydrogen-bonding NH groups. In addition, υC=O at (1745, 1730 cm-1) is a diazetidine ring, υC=O at (1650, 1641 cm-1) is an amide ring, at (3136, 3102 cm-1), the -CH segment of the diazetidine ring. 1H-NMR results show that the characteristic single peak of the imine proton has disappeared, while the (-CH) proton of the diazetidine ring is at (6.96-6.99) (δ) was increased. 13C-NMR shows that the characteristic single peak of imine carbon disappears, the (-CH) peak of diazetidinine is at (61.86-66.82) (δ), and the carbonyl peak of diazetidinine is at (152 –161) (δ).
A genetic algorithm called GOLD was used to dock flexible ligands to protein binding sites, predicting the optimal molecular interaction between the expected compound (M4a-e) and the active binding site of the EGFR protein.24 The PLP fitness scores rank inhibitory activities, and docking studies suggest that all predicted compounds have excellent binding energies with active receptor pockets, potentially showing promising activity with EGFR proteins. The preliminary molecular docking perfectly correlated with subsequent cytotoxicity (in-vitro) studies for compounds (M4a-e) against lung cancers. Amino acids of the epidermal growth factor receptor (PDB: 1M17) interacted with the novel compounds through hydrogen and short contact bonds, including VAL 702, LYS 721, ASP831, THR830, LEU820, MET769, LEU768, GLY772, THR766, LEU764, CYS773, CYS721, and CYS 773. All anticipated final compounds exhibited higher binding energies than the standard drug erlotinib, ranging from (84.70) to (92.77), with a PLP fitness value of (76.20) as shown in Table 2.
The use of in silico tools to predict the pharmacokinetic properties of potential drug candidates during the lead generation and optimization stages has been shown to increase the chances of surviving the high turnover rates of drug discovery. To expedite the drug discovery process, efforts have been made to integrate pharmacokinetic and developmental considerations early in the research process, with a focus on identifying compounds with greater potential to optimize binding.25 All predicted compounds had a bioavailability of 0.55 and a TPSA of less than 140 A0, except compounds M4c and M4e, which had a bioavailability of 0.17, indicating that they entered the systemic circulation. These compounds do not cross the blood-brain barrier (BBB). In addition, none of the compounds had an affinity for P-gp, the protein responsible for preventing chemotherapy drugs from being taken up by cells. Since these compounds are P-gp non-substrates, they are not excreted from the cell by efflux transporters.26 In addition, all predicted compounds complied with the Lipinski rule of five, with the exception of M4c and M4e, which had two violations, as shown in Table 3.
Based on the anti-proliferative evaluation27 on the A549 cell line, we found that the compounds M4c and M4e had outstanding and promising anticancer activity against this cell line for the treatment of lung cancer with IC50 values of 1.75 and 2.05 μm, respectively, after 72 hours of administration, they are more active than the reference product erlotinib with IC50 (11.5) μm after 72 hours. The results of the cytotoxicity studies of the terminal compounds (M4c and M4e) were consistent with the predictions of the molecular docking studies. All synthesized compounds showed excellent anti-proliferative activity except compound (M4d) with IC50 (11.33) μm comparable to erlotinib (11.5) μm and compound (M4b) with slightly lower IC50 (14, 01) μm, higher than that of erlotinib as shown in Figure 2, and A549 cells were also observed by microscopy after 48 hours after exposure to 62.5 μm synthetic compounds (M4a-e). Morphological changes, compared with A549 cells (control cells) in Figure 3.
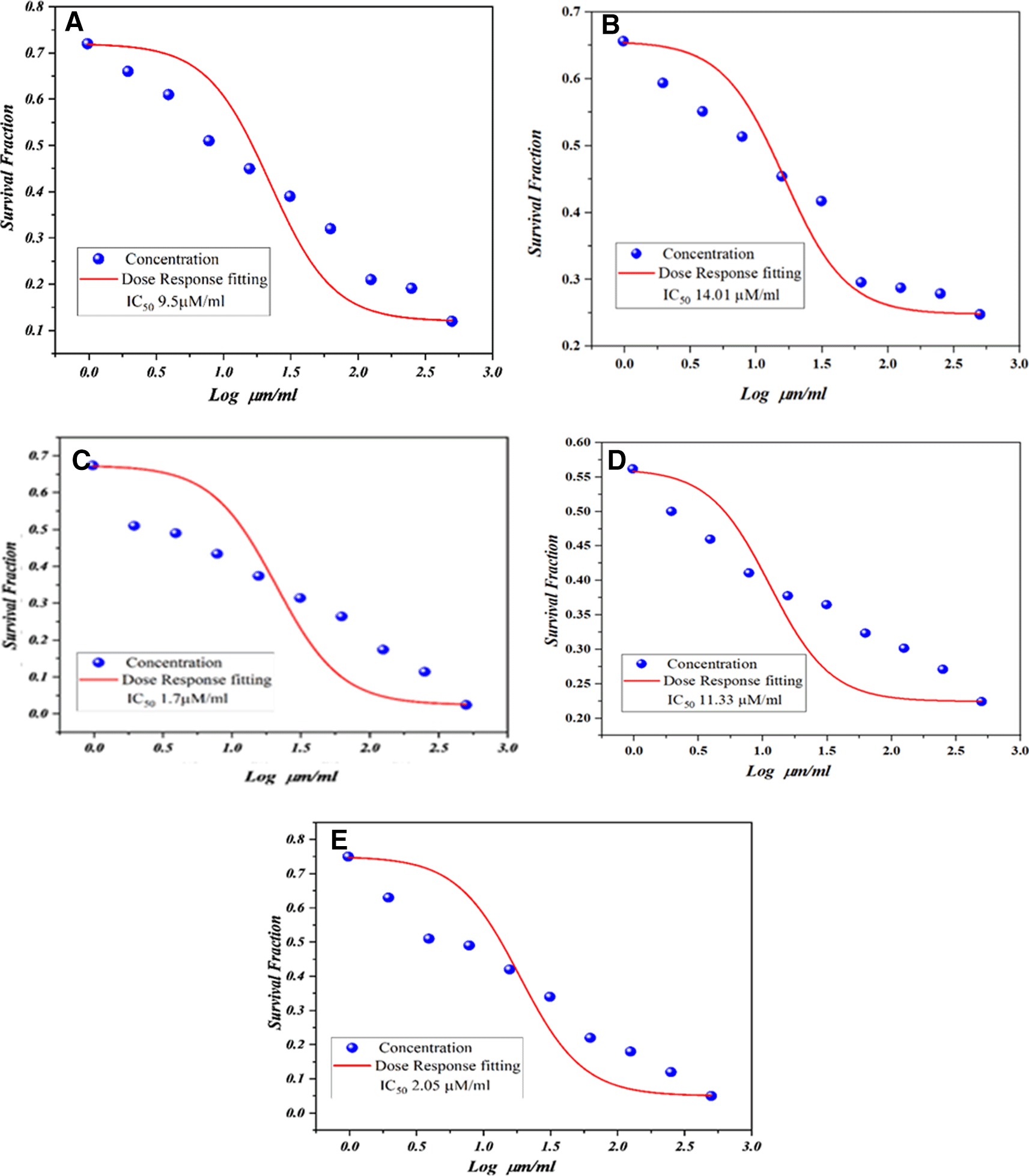
(a)=M4a, (b)=M4b, (c)=M4c, (d)=M4d, (e)=M4e.
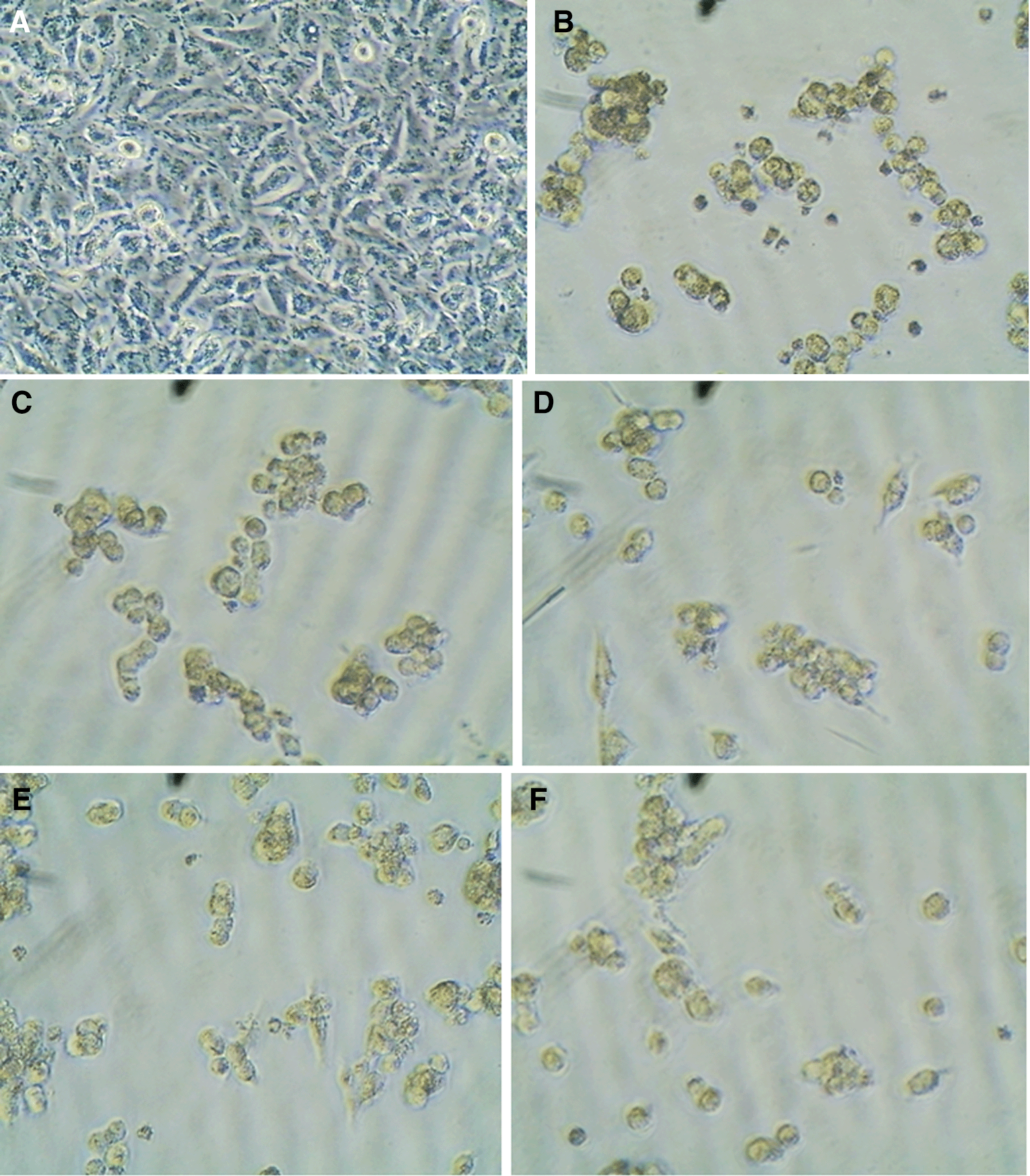
a: control, b: M4a, c: M4b, d: M4c, e: M4d, f: M4e.
A series of novel 1,3-diazetidin-2-one derivatives have been successfully characterized and evaluated using in silico methods including ADME studies. The results showed that all compounds, were strongly passively absorbed from the gastrointestinal tract and conformed to Lipinski's "rule of five except compounds M4c and M4e, with two violations. Molecular docking studies of the final compounds (M4a-e) revealed a significant interaction with the EGFR protein, which outperformed the reference drug erlotinib. The anti-proliferative activity of the synthesized compounds against the lung cancer cell line A549 was evaluated. All synthesized compounds showed excellent anti-proliferative activity except compound M4d, which had an IC50 value (11.33) μm comparable to that of erlotinib (11.5) μm and compound M4b exhibited a slightly higher IC50 than erlotinib after 72 hours. New Compound M4a-e shows excellent correlation between cytotoxicity and molecular docking studies.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)