Keywords
Rhodanine, lung cancer, EGFR mutation.
Rhodanine, lung cancer, EGFR mutation.
Cancer remains one of the main health issues globally.1 As reported by the World Health Organization (WHO), cancer is the most significant cause of death in the world, accounting for over 10 million fatalities in 2020 and by 2040, 29.5 million additional cancer cases are expected to be reported. Lung cancer can be classified into two types2,3: Non-small cell lung cancer (NSCLC) which accounts for 80% to 85% of all lung cancers,4–7 and SCLC (small cell lung cancer) which accounts for 10–15% of all lung malignancies.8 This kind of lung cancer grows and spreads more rapid than NSCLC. Tobacco smoke exposure causes a high mutations and is associated with frequent DNA duplicaions and deletions.9–11 The existence of gene mutations or rearrangements is determined by molecular analysis; these molecular changes include epidermal growth factor receptor (EGFR),12,13 gene mutations, anaplastic lymphoma kinase (ALK) gene rearrangements.14,15 First-generation EGFR tyrosine kinase inhibitors (TKI) that bind to the kinase domain of EGFR reversibly, such as gefitinib and erlotinib, are frequently used to treat NSCLC patients with activating EGFR mutations.16–23 gefitinib/erlotinib resistance has emerged in more than half of previously treated tumors.24,25
Heterocyclic compounds are often employed in medicinal chemistry and are crucial to the pharmaceutical industry’s search for novel bioactive molecules. Rhodanine and its derivatives 2, 4-thiazolidinedione (TZD), whose structure includes a thiazolidine nucleus and a carbonyl group on the fourth carbon, are examples of five-membered heterocyclic compounds having a variety of biological functions (Figure 1).26,27

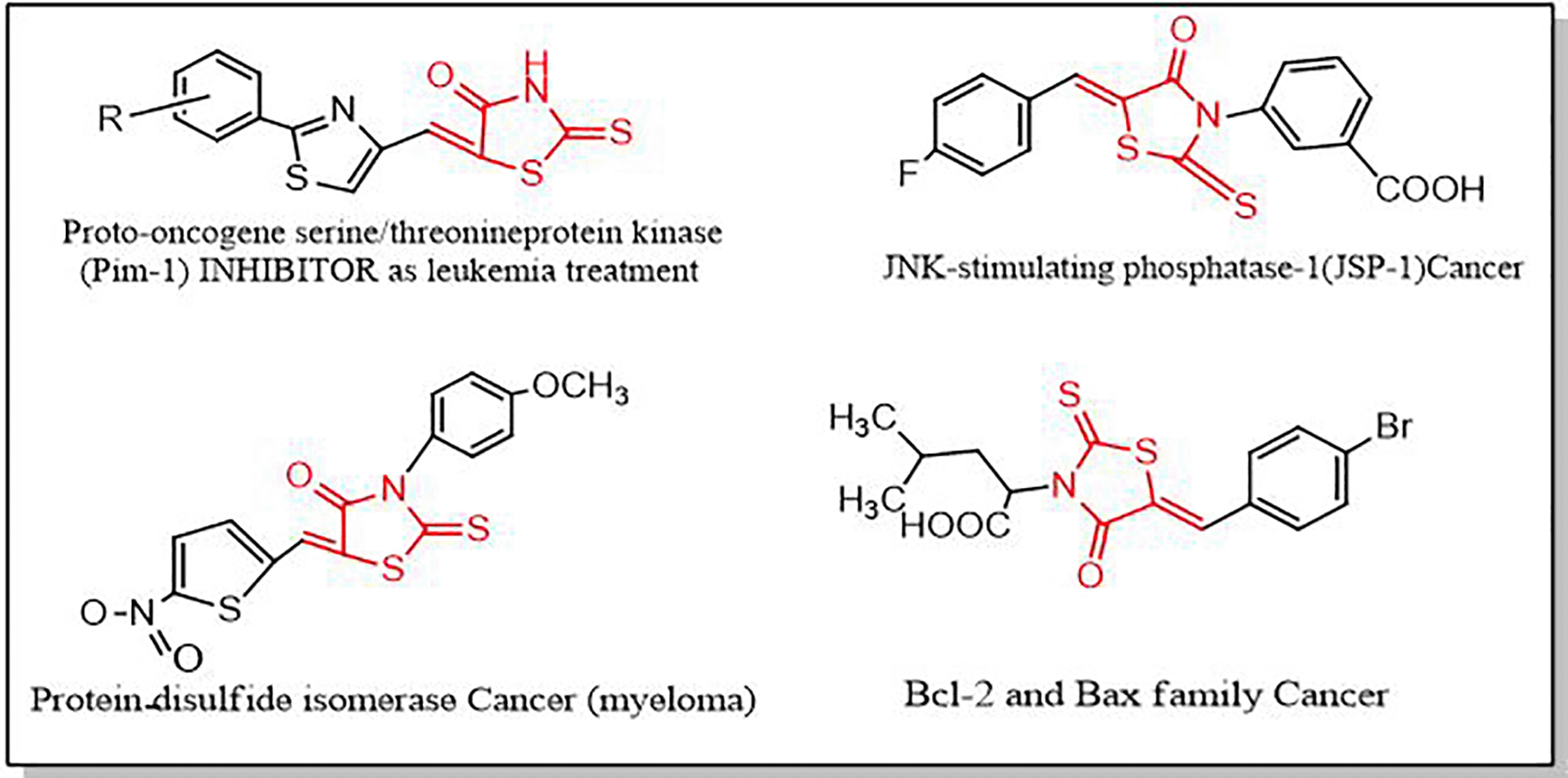
Rhodanine has been recognized as an important type of heterocyclic chemicals in the development of drugs in the last few years.28–30 Several natural and synthesized biological active substances having a rhodanine core are employed as therapeutic treatments, such as anti-inflammatory,31 anti-bacterial,32 anti-tuberculosis,33 anti-diabetic,34 hypnotic,35 anti-parasitic,36 anti-HIV37 and carbonic anhydrase inhibitor agents.38 Anti-cancer properties of rhodamine-containing derivatives are shown in Figure 2.39 Furthermore, the complex-rich chemistry of rhodanine, as well as the significance of heterocyclic compounds, support the future development of synthetic techniques in the chemical industry.40,41
In order to offer higher efficacy of treatment, and to pass the resistance to drug resulting from mutations or inappropriate binding, more effective treatments are required. Moreover, little research has been conducted on the synthesis of amino-rhodanine derivatives. For these purposes, in the present study we synthesized and analysed the anticancer activity of novel N-substituted rhodanine derivatives which are of varying type.
Positions 3 and 5 in the rhodanine ring are highly reactive and play an important role in the design as well as the creation of new drug-like compounds.46,47 So, in the synthesis of N-substituted rhodamine, the regioselectivity of reaction is important to first obtain N-substituted compound, which occurs due to varying factors such as temperature, duration, catalysts and solvents to get the best reaction conditions. We started with 3-amino-2-thioxothiazolidin-4-one, which is commercially available and aldehydes, as shown in Figure 1, the reaction is region-selective at position 3 of 3-aminorhodanine and involves Schiff base formation (Scheme 1). The ideal parameter for the reaction to obtain compound 1a is using acetic acid as a catalyst and using methanol as a solvent, while the high yield of compound 1b can be obtained without using any catalyst.1,48,49

Compounds 2a1-2, 2b1-2 were synthesized via Knoevenagel condensation between aldehyde and the final stages of the first step (1a, 1b), which are N–substituted rhodanine, using NH4OH/NH4Cl as a catalyst and reflux for 6-8 hrs. Condensation in alcoholic solution in an environment of NH4OH/NH4Cl always resulted in 5-[(aryl) alkylidene]-3-aminorhodanine50; Petlichnaya and colleagues obtained similar results (1967 and 1970).48,49 In the synthesis procedure, compounds were highly affected by increasing heat, so the used reflux temperature was not more than 50°C- 60°C.
The structures of the resulting products were examined using spectral data analysis; the Fournier transform infrared (FTIR) spectrum of compounds 1a and 1b showed characteristic absorption bands at 1721 cm-1 and 1737 cm−1, respectively, corresponding to the C=O group of thioxothiazolidin ring. The C=N stretching bands appeared at 1605 cm−1for compound 1a and at 1614 cm−1for 1b. Another two bands appeared at 1418 cm−1 for 1a as well as 1545 cm−1and 1467 cm−1 for 1b, which were attributed to C=C stretching aromatic. Moreover, C=S group appeared at 1104 cm−1 and 1084 cm−1 for 1a and 1b compounds, respectively.
The 1H-NMR spectrum identified a proton resonating as a singlet at δ = 7.83 ppm for 1a and 8.58 ppm for 1b for azomethine group (CH=N), which confirmed the imine group formation. The most significant singlets at δ = 4.24 ppm and 4.37 ppm for compounds 1a and 1b, respectively, correspond to the 2H proton at C5 of thioxothiazolidine ring, explaining the reaction that occurs in 3-amino position of 3-aminothioxothiazolidine core and formation of Schiff base. In compound 1a doublet signals appeared at 7.59 ppm for H-2, 6 and 8.75 ppm for H-3, 5 of the pyridine ring. 1H-NMR (proton nuclear magnetic resonance) spectrum of 1b showed signals of furan ring as doublate of doublat for H-4, doublate for H-5 and doublate for H-3 at 6.81, 7.39 and 8.10 ppm, respectively.
The 13C-NMR analysis identified Schiff base carbon which appeared at 158.2 ppm and 147.5 ppm for 1a and 1b compounds, respectively. The thioxothiazolidine ring C5 resonated at 33.8 ppm in 1a and 35.6 in 1b, and the carbonyl groups appeared at 164.0 ppm and 170.1 ppm for 1a and 1b, respectively. In contrast, the C=S group resonated at 188.1 ppm for 1a and at the 197.8 ppm for 1b. The 13C-NMR spectrum demonstrated another distinctive signal such as 124.2 ppm for C-2, 6, 140 ppm for C-1 and 151.1 ppm for C-3, 5 which represent the carbon atoms of pyridine ring in 1a compound, while 113.6 ppm for C-4, 121.8 ppm for C-5, 148.8 ppm for C-3 and 158.7 ppm for C-1 which refer to carbon atoms of furfural in compound 1b.
The FTIR spectrum of compounds 2a1 and 2a2 showed characteristic absorption bands at 3045 cm−1 and 2933 cm−1 corresponding to =C-H sp2, -C-H sp3 of new compound 2a1 whereas compound 2a2 showed these peaks in the 3174 cm−1 and 3037 cm−1 regions. Two bands at 1737 and 1678 cm−1 corresponded to the C=O of thioxothiazolidin ring in compounds 2a1 and 2a2, respectively. FTIR spectra also showed distinctive peaks in 1593 and 1595 cm−1 regions, corresponding to C=N of new compounds 2a1 and 2a1, respectively. A C=S group appeared at 1097 cm−1 for 2a1 and at 1170 cm−1 for 2a2.
The 1H-NMR spectrum of compounds displayed the most important singlets at the 8.12 ppm for 2a1 and 8.17 ppm for 2a2 of the azomethine group (-N=CH). Also, signals representing the α-hydrogen appears as singlets at 7.94 and 7.99 ppm for 2a1 and 2a2, respectively.
The 13C-NMR (carbon nuclear magnetic resonance) spectra for the same derivatives showed the most significant peaks at 143.4 ppm of C-α for 2a1 and 141.2 ppm for 2a2. Also, compound 2a1 showed 153.6 ppm for –N=CH carbon while this signal appeared at 158.2 ppm for 2a2 derivative.
The FTIR spectrum of compounds 2b1 and 2b2 show peaks in 2951, 2841, 1703, 1591 and 1081 cm−1 for compound 2b1 corresponding to =C-H sp2, -C-H sp3, C=O, C=N and C=S while these signals appeared at 3120, 2918, 1710, 1610 and 1057 cm−1 in 2b2.
The 1H-NMR spectrum identified protons resonating as singlet at 8.14 ppm for compound 2b1 and at 8.26 ppm for compound 2b2 for the azomethine group (-N=CH) which confirmed the imine formation. Another distinct 1H-NMR signal appeared as a singlet at 7.68 ppm and 7.58 ppm represents the α-hydrogen of 2b1 and 2b2, respectively.
The 13C-NMR spectra for 2b1 and 2b2 showed the most significant peaks at 143.3 ppm of C-α of methine group for 2b1 and 142.6 ppm for 2b2. Also, compound 2b1 showed a signal at 132.9 ppm for –N=CH carbon, while this signal appeared at 134.5 ppm for 2b2 derivative.
As a result, all of these distinguished bands and signals identify the functional groups present in each particular derivative and explain the formation of new scaffolds.
Docking of co-crystallized ligands was performed, and the most active ligand was shown to be at the receptors’ expected catalytic active region. As shown by Figure 3, both compounds 2a2 and 2a1 occupied the same binding site as erlotinib.

Compound 2a2 coordinated with THR766 amino acid of receptor with a binding score of 2.856 Å and the estimated docking scores are shown in Table 1. The score showed a high affinity of 2a2 and a binding plp fitness of 72.71 Kcal/mmol. This result made the ligand more active against their receptor compared to standard inhibitor erlotinib. Moreover, ligand 2a1 showed good binding to the receptor compared with the standard ligand as clarified in Figure 3. It showed a 70.29 Kcal/Mol plp fitness and the binding involved two hydrogen bonds with THR766, THR830 with the amino acid of the receptor protein. While the docked poses of compound 2b2 showed a binding affinity slightly greater than the standard at about 68.88 Kcal/Mol plp fitness and 2.947 Å. On the other hand, compound 2b1 exhibited lower values than Erlotinib plp fitness with a binding score 2.696 Å of the receptor. The optimal position of the ligand was determined to be the best-scored ligand into catalytic active sites. Thus, the synthesized rhodanine derivatives appeared to be capable to access the target’s active site and position (through specific connections) into an optimal state for nucleophilic attacks by the amino acid’s (THR766, THR830) residue of the EGFR receptor.
The anti-tumor efficacies of the synthesized compounds 2a1, 2a2, 2b1 and 2b2 were examined in vitro using an A549 cell line. The response was measured at 24, 48 and 72 h. It is described in Table 2 and Figures 4-6. The responses were different at different times: some compounds showed an optimal effect during the first 48 h, (2a1 and 2a2), while another compound took more time to have a high anti-tumor effect (2b1). All compounds were examined using the same cancer cell line (A549). As shown in Figure 4, compound 2a2 (IC50 10.8 μg/mL) was much more potent in inhibiting lung cancer cells than erlotinib during the first 24 hours. Furthermore, as seen in Figure 5, compound 2a1 exhibited excellent anticancer activity at 48 hours and was more potent than erlotinib with IC50 (32.59 μg/mL), with a higher safety when its cytotoxicity was examined on HdFn normal cell and showed an IC50 of about 154.4 μg/mL, which made it safer than erlotinib with an IC50 of 122.8 μg/mL. Also, after examination on human normal cell line, compound 2a1 showed high antitumor activity at 72 h and excellent safety as displayed in Figure 6.

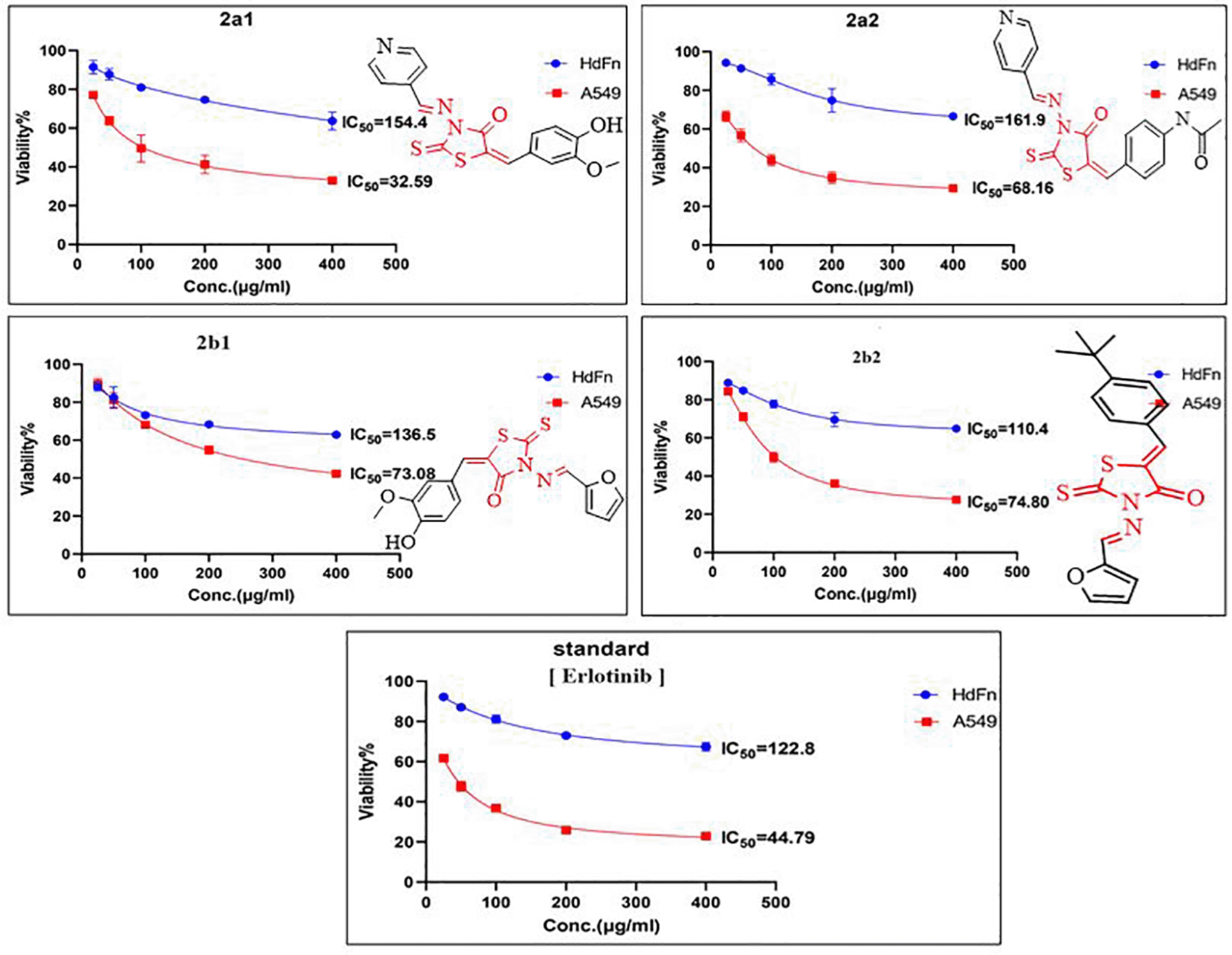

In contrast, other compounds such as 2b1 and 2b2 were less efficient on the cancer cell line (A549). Thus, they showed a moderately lower IC50 value (67.57 μg/mL and 72.02 μg/mL, respectively) with their optimal activity starting after 72 hours. In terms of safety, compared with erlotinib, these two compounds exhibited high safety in their cytotoxicity examination on HdFn normal cell Figure 6. These examinations results indicated significant outcomes for rhodanine derivatives, which might become a candidate for lung cancer treatment.
All chemicals and solvents were commercially available from Sigma-Aldrich or Merck. FT-Infrared spectra were analyzed by 8400 Shimadzu KBr spectrophotometer, 1H-NMR and 13C-NMR (300 and 75 MHz) spectra were analyzed using a Bruker spectrometer. The melting points were measured without correction using an electro-thermal melting point apparatus. The reaction progress was monitored by thin-layer chromatography (TLC, 0.25 mm-thick precoated silica Merck plates).
General synthesis of N-substituted-rhodanine derivatives (1a and 1b)
To a solution of 3-amino-2-thioxothiazolidin-4-one (1mmol) in 2.5 mL methanol, aldehydes (1.2 mmol) in 1.5 mL methanol were added slowly. The reaction mixtures were stirred at room temperature with glacial acetic acid as a catalyst for compound 1a and without any catalyst for compound 1b for 4 to 6 h, and were monitored by TLC. After that, the two mixture products were recrystallized from methanol. After recrystallization, N-substituted-rhodanine derivatives were obtained as follows:
(E)-3-((pyridin-4-ylmethylene) amino)-2-thioxothiazolidin-4-one (1a)

The product was obtained as an off-white powder (200 mg, 84% yield); m.p: 168°C-170°C; Chemical formula: C9H7N3OS2. FTIR (KBr, cm−1) vmax: 3296 (=C-H sp2 stretching), 2814 (-C-H sp3 stretching), 1721 (C=O stretching), 1605 (C=N stretching), 1418 (C=C aromatic stretching), 1293 (C=S stretching), 1208 (=C-N stretching), 1104 (C=S stretching), 1063 (-C-N stretching), 869 (C-S stretching) 784 (C-H aromatic out of plane bending). 1H-NMR (300 MHz, DMSO-d6) δ ppm 4.24 (s, 2H, CH2), 7.59 (d, 2H, H-2, 6), 7.83 (s, 1H, CH=N), 8.75 (d, 2H, H-3, 5). 13C-NMR (75 MHz, DMSO-d6): 47.2 (CH2), 124.2 (C-2, 6), 140.3 (C-1), 151.1(C-3, 5), 158.2 (CH=N), 164.0 (C=O), 188.1 (C=S).
(E)-3-((furan-2-ylmethylene) amino)-2-thioxothiazolidin-4-one (1b)

The product obtained was a yellow glittery crystal (165 mg, 73% yield); m.p: 130°C-132°C; Chemical formula: C8H6N2O2S2. FTIR (KBr, cm−1) vmax: 3128 (=C-H sp2 stretching), 2928 (-C-H sp3 stretching), 1737 (C=O stretching), 1614 (C=N stretching), 1545 and 1467 (C=C aromatic stretching), 1390 (C=S stretching), 1228 (C-O stretching), 1084 (C=S stretching), 1016 (C-N stretching), 825 (C-S stretching), 767 (C-H aromatic out of plane bending). 1H-NMR (300MHz, DMSO-d6): δ 4.37 (s, 2H, CH2), 6.81 (dd, 1H, H-4), 7.39 (d, 1H, H-5), 8.10 (d, 1H, H-3) 8.58 (s, 1H, CH=N). 13C-NMR (75 MHz, DMSO-d6): 35.2 (CH2), 113.6 (C-4), 121.8 (C-5), 147.5 (CH=N), 148.8 (C-3), 158.7 (C-1), 170.1 (C=O), 197.0 (C=S).
Synthesis of 5-substituted rhodanine derivatives 2a1, 2a2 and 2b1, 2b2 (final compounds)
To a warm solution of 1 mmol of compound (1a or 1b) in 3 mL methanol, benzaldehyde derivatives (1.2 mmol) were dissolved in 2 mL methanol mixed with 0.2 mL. NH4OH and 0.1g of NH4Cl dissolved in 0.2 mL water were added slowly and refluxed at 50°C with stirring for 6-9 h.48 The reaction mixture was monitored by TLC. The products of 5-substituted rhodanine derivatives recrystallized using a mixture of water and methanol and were obtained as follows:
(E)-5-(4-hydroxy-3-methoxybenzylidene)-3-((E)-(pyridin-4-ylmethylene)amino)-2-thioxothiazolidin-4-one (2a1)

The product obtained was a brown solid (300 mg, 83% yield); m.p: 105°C-107°C; Chemical formula: C17H13N3O3S2. FTIR (KBr, cm−1) vmax: 3045 (=C-H sp2 stretching), 2933 (-C-H sp3 stretching), 1737 (C=O stretching), 1593 (C=N stretching), 1512 (C=C aromatic stretching), 1406 (C-O stretching), 1278 (=C-N stretching), 1097 (-C-N and C=S stretching), 812 (C-S stretching). 1H-NMR (300 MHz, DMSO-d6) δppm 3.82 (s, 3H, OCH3), 4.91 (s, 1H, OH), 7.02 (d, 1H, H-5′), 7.38 (s, 1H, H-2′), 7.42 (d, 1H, H-6′), 7.94 (s, 1H, H-α), 7.97 (d, 2H, H-2, 6), 8.12 (s, 1H CH=N), 8.30 (d, 2H, H-3, 5).13C-NMR (75MHz, DMSO-d6) δppm 56.2 (OCH3), 111.0 (C-2′), 115.6 (C-5′), 115.8 (-C= of thioxothiazolidin ring), 115.9 (C-2, 6), 122.8 (C-6′), 129.7 (C-1′), 143.39 (C-α), 148.4 (C-1), 148.6 (C-3′), 149.3 (C-3, 5), 150.4 (C4′), 153.6 (CH=N), 166.5 (C=O), 191.5 (C=S).
N-(4-((E)-(4-oxo-3-((E)-(pyridin-4-ylmethylene)amino)-2-thioxothiazolidin-5-ylidene)methyl)phenyl) acetamide (2a2)

The product obtained was a dark brown solid (300 mg, 78% yield); m.p; 98°C-100°C; Chemical formula: C18H14N4O2S2. FTIR (KBr, cm−1) vmax: 3174 (=C-H sp2 stretching), 3037 (=C-H sp3 stretching), 1678 (C=O stretching), 1595 (C=N stretching), 1523 (C=C aromatic stretching), 1410 (C=S stretching), 1317 (=C-N stretching), 1170 (C=S stretching), 1093 (C-N stretching), 833 (C-S stretching). 1H-NMR (300 MHz, DMSO-d6) δppm 2.08 (s, 3H, CH3), 7.38 (s, 1H, N-H), 7.61 (d, 2H of H-3′, 5′), 7.71 (d, 2H of H-2′, 6′), 7.84 (d, 2H of H-2, 6), 7.99 (s, 1H of H-α), 8.17 (s, 1H, CH=N), 8.43 (d, 2H of H-3,5). 13C-NMR (75 MHz, DMSO-d6) δppm 24.6 (CH3), 119.0 (-C= of thioxothiazolidin ring), 121.6 (C-2, 6), 122.6 (C-3′, 5′), 128.4 (C-2′, 6′), 129.3 (C-1′), 139.8 (C-4′), 141.2 (C-α), 142.5 (C-1), 150.4 (C-3, 5), 158.2 (CH=N), 169.0 (C=O), 169.6 (C=O of acetamid group), 192.0 (C=S).
(E)-3-((E)-(furan-2-ylmethylene)amino)-5-(E)-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (2b1)

The product obtained was a brown solid (300 mg, 83% yield); m.p: 85°C-87°C; Chemical formula: C16H12N2O4S2. FTIR (KBr, cm−1) vmax: 2951 (=C-H sp2 stretching), 2841 (-C-H sp3 stretching), 1703 (C=O stretching), 1591 (C=N stretching), 1460 and 1431 (C=C aromatic stretching), 1381 (C=S stretching), 1290 (C-O stretching), 1081 (C=S stretching), 1022 (C-N stretching), 819 (C-S stretching). 1H-NMR (300 MHz, DMSO-d6) δppm 3.83 (s, 3H, OCH3), 5.94 (s, 1H, OH), 6.77 (d, 1H, H-6′), 7.04 (dd, 1H, H-4), 7.27 (d, 1H, H-5), 7.48 (d, 1H, H-5′), 7.60 (s, 1H, H-2′), 7.68 (s, 1H, Hα), 7.94 (d, 1H, H-3), 8.14 (s, 1H, CH=N). 13C-NMR (75 MHz, DMSO-d6) δppm 56.2 (OCH3), 112.8 (C-2′), 113.3 (C-4), 115.6 (-C= of thioxothiazolidin ring), 116.0 (C-5′), 120.2 (C-5), 122.8 (C-1′), 126.0 (-C= of C-6′), 132.9 (CH=N), 143.3 (Cα), 145.4 (C-3), 148.5 (C-4′), 149.2 (C-1), 150.5 (C-3′), 169.6 (C=O), 191.4 (C=S).
(E)-5-(4-(tert-butyl)benzylidene)-3-((E)-(furan-2-ylmethylene)amino)-2-thioxothiazolidin-4-one (2b2)

The product obtained was a brown solid (250 mg, 70% yield); m.p: 112°C-114°C; Chemical Formula: C19H18N2O2S2. FTIR (KBr, cm−1) vmax: 3120 (=C-H sp2 stretching), 2918 (-C-H sp3 stretching), 1710 (C=O stretching), 1610 (C=N stretching), 1541(C=C aromatic stretching), 1232 (C-O stretching), 1057 (C=S stretching), 1018 (C-N stretching), 817 (C-S stretching). 1H-NMR (300 MHz, DMSO-d6) δppm 1.33 (s, 9H, 3×CH3), 5.42 (t, 1H, H-4), 7.11 (d, 1H, H-5), 7.47 (d, 2H, H-3′,5′), 7.58 (s, 1H, Hα), 7.67 (d, 2H, H-2′,6′), 7.89 (d, 1H, H-3), 8.26 (s, 1H, CH=N). 13C-NMR (75 MHz, DMSO-d6) δppm 31.3 (3×CH3), 35.2 (C (CH3)3), 113.1 (C-4), 117.4 (C= of thioxothiazolidin ring), 118.1 (C-5), 126.2 (C-3′, 5′), 128.5 (C-2′, 6′), 133.8 (C-1′), 134.5 (CH=N), 142.6 (Cα), 144.4 (C-3), 149.4 (C-1), 150.1 (C-4′), 169.4 (C=O), 196.0 (C=S).
All synthesized compounds were analyzed for their activity against lung tumor using a MTT(3-(dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide) assay on A549 lung cancer cell line. The cytotoxic effect of the newly synthesized final compounds were also examined on HdFn normal cells to determine their safety on human normal cells. The antitumor activity and safety of these compounds were compared with clinically used antitumor (TKI) drug erlotinib, which is used as a standard reference. The evaluation of 3-aminorhodanine derivatives was carried out using different concentrations from 25 μg/mL to 400 μg/mL.51 The inhibitory concentrations (IC50, μg/mL) were measured at 24, 48, and 72 h after application of our new compounds (2a1-2, 2b1-2) and compare the viability of cancer cells within non treated normal cell.
The molecular docking study was performed using CCDC (Cambridge Crystallographic Data Centre), fully licensed Genetic Optimization Ligand Docking (GOLD) Suite (v.5.7.1)52,53 The protein (4HJO) of EGFR crystal structure data was obtained from the Protein DataBank.54 The protein structure and ligands were prepared and corrected before the docking process. Preparation was done by removing of all ligands and water molecules that were not involved in the binding site. energy was calculated and minimized using MM2 minimize and MM2 dynamic option. The receptor was compiled before the docking process by adding polar hydrogen atoms to achieve specific ionization and tautomeric positions of amino acid residues. The binding sites were identified using the co-crystallized ligand erlotinib. All of the protein residue characterized within 10 A° of the typical ligands for the docking process. The piecewise linear potential (plp) function was used in the scoring function. Finally, the output was downloaded as a mol.2 file.
In the docking study site scores were computed to assess whether binding ligands are catalytically activated sites, and calculating the binding energies of novel compounds with the protein (EGFR), including the H-bond, in addition to other bonds that ligands developed with the receptor amino acids in the active site.
A new series of rhodanine derivatives consisting of significant aromatic groups, 2a1-2 and 2b1-2, were designed, synthesized and investigated for their potential anti-tumor effects. Some of these new rhodanine derivatives showed the maximum anti-tumor effect at 200 μg/mL concentration against A549 lung cancer cell line. However, rhodanine derivatives that contained pyridine were more efficient toward lung cancer cells; this result was confirmed by the in vitro anti-tumor analysis and docking studies, in which the computed scores revealed that anticipated binding sites displayed catalytic active site features and that these compounds had an anti-tumor effect when compared to the reference medication erlotinib, which was classified based on the plp fitness.
Compounds 2a1 and 2a2 were more potent than standard erlotinib and showed excellent activity at different times. Thus, the new synthesized rhodanine compounds showed a high potential for use as innovative therapeutic agents in the treatment of lung cancer.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)