Keywords
Norfloxacin , nanoemulsion , self nano-emulsifying drug delivery system (SNEDDS)
Norfloxacin , nanoemulsion , self nano-emulsifying drug delivery system (SNEDDS)
Self-emulsifying drug delivery systems (SEDDS) are isotropic homogeneous pre-concentrate mixes of oil, surfactant, co-surfactant, and drug that, when gently agitated in an aqueous media, rapidly produce fine oil-in-water (o/w) emulsions.1 SEDDS improve the oral bioavailability of the medicine by enhancing its solubility and keeping it spread in small globules of oil during its passage through the gastrointestinal system.2 Also, bioavailability will be improved as a result of avoiding the first pass effect that when bile acids are present, lingual and pancreatic lipases activate on the oily phase of the SEDDS, resulting in the creation of emulsified mono-glycerides, diglycerides, and fatty acids and the generation of intestinal mixed micelles. As these mixed micelles travel through the enterocytes, they produce chylomicrons, they can route the medication into lymphatic pathways instead of blood vessels.3 Because the diameter of the emulsion droplets generated ranges from nanometers to several microns, self-emulsify drug delivery systems are classified as self-nano-emulsifying (SNEDD) and self-micro-emulsifying (SMEDD).4
Norfloxacin (NOR) is a second generation fluoroquinolone. This broad-spectrum antibacterial drug is widely used to treat a variety of infection disorders involving the gastrointestinal or genitourinary systems.5,6
Norfloxacin’s chemical name is 1-ethyl-6-fluoro-4-oxo-7-piperazin-1-yl-1H-quinoline-3-carboxylic acid. It has a very low solubility in water (0.28 mg/mL), and its color is a very light yellow. NOR is very basic in nature (pKa 6.39) and has an empirical log P value of 1.47. Additionally, the clinical effectiveness of this BCS class IV medication is affected by its poor bioavailability (30%), protein binding value (15%), and about 30% of drug excreted unaltered.7 There are a few attempts to improve the pharmaceutical properties of NOR such as dispersible tablets, extended-release matrix tablets, gastro-retentive floating tablets, cyclodextrin-containing suspension, solid lipid nanoparticles, suspensions, gastro-retentive floating microspheres, and sustained release alginate micro-beads.
The goal of this project is to develop norfloxacin as an oral self-emulsifying liquid formulation that will improve solubility and thus bioavailability.
Norfloxacin, coconut oil, mineral oil, olive oil, corn oil, spearmint oil and castor oil from Hangzhou Hyper chemicals (China). Oleic acid, clove oil and methanol were purchased from G.C.C., (UK). PEG 400, propylene glycol were obtained from CDH (India). PEG200, tween (20 and 80), span (20 and 80) were obtained from Thomas baker (India).
The saturated solubility of norfloxacin in various oils, surfactants, and co-surfactants was determined. The oils that were used were (coconut oil, mineral oil, olive oil, oleic acid, corn oil, spearmint oil, clove oil, castor oil). Surfactants and co-surfactants (tween 20, 80, span 20, 80) (PEG200, 400, PG). In order to assess the solubility, an excess amount of norfloxacin powder was added to 5ml of each excipient (oil, surfactant, and co-surfactant) contained in small, firmly sealed glass tubes. After 3 days in a shaking water bath at (25±0.5)°C, the tubes were centrifuged at a rate of 3000 rpm for twenty minutes, and the supernatant layer was filtered with a 0.45 m syringe filter. Methanol was used to dilute the samples. A UV-visible spectrophotometer (Biotech Engineering Management Co) is utilized in order to determine solubility. The measurement was carried out in triplicate.8,9
Pseudoternary phase diagrams for oil (oleic acid), surfactant (tween20) and co-surfactant (PEG200) represented as Smix were developed. Water was obtained at room temperature using the water titration technique, and each of them represents a side of the triangle. Several Smix (1:1, 1:2, 1:3, 1:4 and 2:1, 3:1, 4:1) ratios of surfactant and co-surfactant were utilized, and oil to particular Smix ratios (ranging from 0.5:9.5 to 9.5:0.5) were prepared.10,11
To manufacture norfloxacin liquid self-emulsifying drug delivery (SEDD) formulations, a vortex mixer was used to combine norfloxacin (400 mg) with oleic acid oil, tween 20, and PEG200 (Smix) in a screw-capped glass vial. The mixture was then warmed in a water bath for half an hour at 50°C to induce homogeneity.12 The (28) produced formulae’ components are shown in Table 1.
The liquid SEDDS formulations were diluted in two separate 1000 ml beaker with deionized water and 0.1 N HCl (50, 100, 250, and 1000 times) to mimic in vivo environments. The diluted systems were shaken with the aid of a magnetic stirrer to ensure complete homogeneity and done at 37°C to simulate body temperature.13
These systems were kept at room temperature for 24 hours before being visually examined for signs of phase separation, drug precipitation, and droplet coalescence.14
Centrifugation test
The SEDDS formulations were centrifuged at 3500rpm for 20 minutes to check for phase separation, creaming, or cracking.15 Following the centrifugation test, the formulations that succeeded will be put to another thermodynamic test.16
Heating-cooling cycle (H/C cycle)
The stabled formulas were subjected to six cycles of heating and cooling at two different temperatures (4°C and 45°C). Formulas would stay at each temperature for at least 48 hours. The formulas that did not show any phase separations or creaming were subjected to freeze-thraw cycles.17
Freeze-thaw cycles
Stable SE formulas were subsequently subjected to freezing and thawing (-20°C and 25°C). They were carried out in three cycles, each lasting 48 hours.18
Self-emulsification time and dispersibility test
The addition of liquid self-emulsify formulas to 500 mL of 0.1N HCl under gentle magnetic stirring was visually observed. Emulsification time was established as the amount of time needed for the prepared formulations to completely disperse and form an emulsion. When a transparent homogenous system was attained. The in-vitro efficiency of the liquid SEDDS formulations was examined visually. It is believed that the creation of clear SEDD occurs best within a minute, since prolonged times result in a milky look.19 As indicated by the grade mentioned in Table 2.20
Using a UV spectrophotometer, the drug content of the chosen formulations (F1, F2, F5, F6, F8, F13) that passed the previously indicated characterisation tests was determined. In a 100 mL volumetric flask, a known amount of norfloxacin-SEDDS was accurately measured. The flask was then filled with methanol and sonicated for around 15 minutes to ensure full drug dissolution. The solution was appropriately filtered via a 0.45-micrometer membrane, diluted, then tested for drug concentration with a UV-spectrophotometer at 278 nm.21–24
The droplet size investigation was carried out using a dynamic light scattering approach, which examines the variance in light scattering caused by particle Brownian motion. Light scattering was measured at room temperature at a 90° angle of laser scattering.25
The liquid norfloxacin-self emulsify formulation was diluted hundred times (0.25mL from each formula in 25 mL) with filtrated distilled water and gently agitated with a magnetic stirrer to generate a fine emulsion. The diluted emulsion was poured in a disposable cuvette in the sample chamber to analyze droplet size and PDI.26
Dissolution apparatus-II was used to establish in-vitro drug release profiles of SEDDS formulae as well as norfloxacin tablet. The medium was 750 ml of acetate buffer pH,4 and the test was carried out using stirring paddles at a speed of 50 rpm and at a temperature of 37°C. The dialysis bag approach was used for drug release investigations to obtain true amounts of free drug without interference with unreleased drug from the formula.27 Aliquots (5 ml) of samples were obtained at 5,10,15,20,30,45,60 minute intervals. To maintain a sink condition, the same quantities of new dissolving medium (ph4) were replaced. The amount of drug dissolved was filtered and measured with a UV-vis spectrophotometer at 278 nm.28
The zeta potential analyzer was used to examine the best liquid SEDD formulation (F1) (Brookhaven instrument). The zeta potential value indicates the repulsive forces between the droplets.29,30 The aqueous dispersion of the produced SE formula was made in the same manner as in the section of globule size measurement.
The solubility of norfloxacin in different oils, surfactants, and co-surfactants was determined as showen in Table 3. To find suitable vehicles with a high solubilizing ability for norfloxacin. The importance of maximum drug solubility is first and mainly an optimization of drug loading capacity. Second, drug precipitation is avoided during dilution, and the final volume of SEDDS is reduced, making it acceptable for oral administration.31 At last, while storage, the whole system’s stability will improve.
The solubility of norfloxacin in different oils is as follows: oleic acid >olive oil >clove oil >castor oil >corn oil > spearmint oil >coconut oil >mineral oil.
As oleic acid had the maximum solubility for norfloxacoin, it was chosen as the oil phase of SEDDS.
The solubility of norfloxacin in various surfactants reveals that the maximum solubility was observed in tween 20 (non-ionic surfactant with HLB values = 16.7),32 which was chosen as the best surfactant.
Norfloxacin solubility was also investigated in several co-surfactants that are critical for interfacial film flexibility and decreasing interfacial tension.33
The medication had a low solubility in PG, but the maximum solubility was seen in PEG200, thus it was chosen.
It was observed from phase diagrams in Figure 1 that increasing the ratio of surfactant to co-surfactant in Smix from 1:1 to 2:1 or 3:1 improved the nanoemulsion shaded area. Moreover, there was a reduction in nanoemulsion area at a Smix 4:1 ratio. The reason for the increased clarity of the emulsions produced as Smix increased was a decrease in the total system’s free energy. Thus, smaller emulsion droplet size reflects a formulation with greater visual clarity. On the other hand, a further increase in Smix may facilitate water breakthrough oil globule, resulting in their disruption and more oil globules released into the aqueous phase.34 In addition, as the co-surfactant (PEG200) concentration increased (1:2, 1:3, and 1:4), the nanoemulsion area decreased in comparison to the 1:1 ratio, which was attributed to the decrease in surfactant (tween 20) concentration from 50% in the 1:1 ratio to 20% in the 1:4 ratio confirming the fact that the surfactant has a primary role by being adsorbed on the surface of oil droplets to provide the essential barrier for the formation of o/w emulsion while the co-surfactant serves as an adjuvant by giving flexibility to the formed barrier film, further lowering the interfacial tension.35 These results concurred with that observed by different studies on self-nanoemulsification.36–38
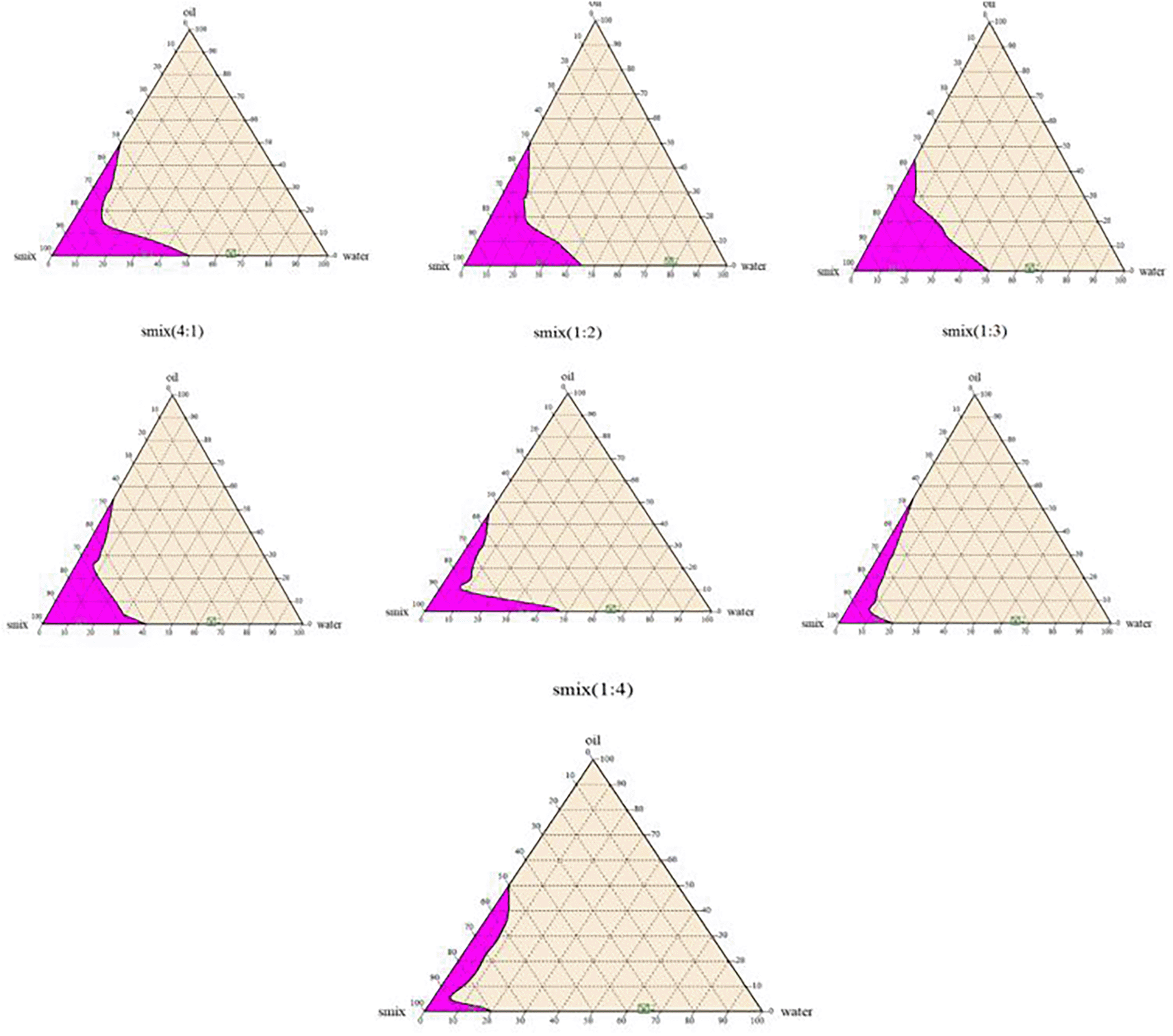
The optimum o/w nano-emulsion area was produced with a low oil: Smix ratio (1:9, 2:8, 3:7, 4:6) because it provided superior stability and a smaller droplet size; this result was consistent with the data.39 As a result, these oil: Smix ratios were chosen for further investigation. Furthermore, raising the oil concentration reduced the nanoemulsion region, which might be due to increased interfacial tension and decreased efficacy of surfactant and cosurfactant to overcome it.
Based on pseudoternary phases diagram,twenty-eight liquid SEDD formulae were produced utilizing various tween 20: PEG200 (Smix) ratios of 1:1, 1:2, 1:3, 1:4, 2:1, 3:1, and 4:1, and oil: Smix ratios of 1:9, 2:8, 3:7, and 4:6. All of the produced formulations underwent a visual assessment of their appearance and other physical characteristics.
The twenty-eight anhydrous formulae that were synthesized had a uniform, clear yellow color and exhibited no evidence of phase separation or drug precipitation.
Robustness to dilution
The SEDDS must be able to resist dilution without exhibiting any phase separation or drug precipitation in order to be employed as a drug delivery vehicle, and diluting SEDDS formulations up to 1000 times with all of the abovementioned diluents can mimic the in-vivo conditions.40 As shown in Table 4, the majority of reconstituted formulations demonstrated good emulsion stability, as evidenced by the preservation of their appearance with no evidence of drug precipitation or phase separation in all dilution media after 24 hr of storage at room temperature. This suggests that such systems may be suitable for oral administration, as they have a high probability of maintaining their emulsified state throughout their passage in the GIT with no phase separation.41
Several formulations failed this test and became turbid and unstable after 24 hours, possibly due to a low concentration of tween 20 in these formulations that if the formulation comprises water-soluble co-surfactant, precipitation is frequent and can be prevented by raising the surfactant concentration.42 In addition, the low concentration of Tween 20 was insufficient to emulsify the high oil content, particularly when it was partially replaced with PEG200. Yet, as the concentration of surfactant rises, both surfactant and co-surfactant exert their functions correctly.
Furthermore, modifying the pH and content of the aqueous phase had no effect on the emulsion characteristics.40
It was also noted that as the concentration of oil increases, the formulation’s stability decreases.43
The SE formulations were visually evaluated after being subjected to centrifugation and abrupt temperature changes, including the heating-cooling cycle and the freezing-thawing cycle.
The observations of the thermodynamic stability investigation for several formulations of norfloxacin SEDDS were displayed in Table 5.
The formulations were visually assessed for signs of phase separation, cracking, or creaming. The results show that most of the formulations passed all three tests and were stable. However, some formulations failed one or more tests, indicating instability. The failed formulations may be prone to phase separation or other forms of instability upon storage or transportation.
The majority of the tested formulae had a short self-emulsification time, demonstrating their capacity to make emulsion efficiently and simply.
Table 6 shows the results of different grades of microemulsion and the measured self-emulsification times created by twenty-eight formulations with varying Smix ratios and oil proportions. The previously indicated visual grading method was utilized to evaluate the in-vitro performance of the tested formulae.
The study of emulsification time and visual observations revealed that six formulations achieved self-emulsification times of 1 minute with a clear appearance and were certified as grade A (the best) liquid SEDDS. It was also discovered that in formulas F1,F8,F15,F22 containing oil: Smix ratios of 1:9, 2:8, 3:7, 4:6 respectively, keeping Smix ratios of 1:1 as the oil to Smix ratio increased, the process of emulsification slowdown, so when fixing Smix and increasing in oil content leads to a gradual increase in emulsification time. However, increasing the concentration of tween 20 resulted in a significant decrease in emulsification time, as well as improved uniformity and bluish color nature of the microemulsion, improving the spontaneity of emulsification.
The ability of surfactants to improve formation of an emulsion and dispersion by lowering the oil-aqueous interfacial tension may explain why higher surfactant concentration was related with shorter self-emulsification time.44
This implies that emulsification efficiency is determined by selecting the appropriate oil ratio as well as the appropriate Smix ratio that provided the optimum emulsification capability, rate, and stability.45
The drug content of the selected formulae is provided in Table 7. The drug content of norfloxacin SEDDS formulations was meeting USP requirements and falling within an acceptable range (85%-115%). These findings indicate that there was no drug precipitation in the developed formulations and that norfloxacin was homogeneous and stable in SEDDS vehicles.46
The experimental results of globule size and PDI measurement for norfloxacin-nanoemulsion in distalled water were presented in Table 8. At a constant Smix ratio, it was discovered that increasing the oil% with a subsequent decrease in the quantity of surfactant and co-surfactant in the mixture resulted in a slight increase in globule size. The explanation might be due to a lack of surfactants and co-surfactants that can localize at the oil/water interface of a droplet to reduce interfacial tension and stabilize the system.47
| Formula code | Particle size (PS) | Polydispersity index (PDI) |
|---|---|---|
| F1 | 57.4±1.730 | 0.351 |
| F2 | 124.2±1.548 | 0.210 |
| F5 | 99.5±1.073 | 0.005 |
| F6 | 86.7±1.781 | 0.395 |
| F8 | 173.1±1.782 | 0.396 |
| F13 | 144.3±1.837 | 0.447 |
Droplet size distribution is an important issue to consider when developing SMEDDS. The lower the PDI value, the narrower the droplet size distribution, indicating uniformity, homogeneity, and mono-dispersion of the formulated self-nanoemulsion, as well as long term stability.48 All SEDDS formulae had low PDI values, indicating that oleic acid globules were dispersed within the formulation and confirmed its homogeneity.34,49–52
Figure 2 depicts the in-vitro release profiles of the produced formulations and the norfloxacin tablet in PH4 dissolving media over 1 hour. At the end of 1 hour, the majority of tested SNEDDS formulae demonstrated a consistently better release rate than the tablet. The rapid spontaneous emulsification capabilities of SNEDDS, as well as the formation of small globules with a high surfactant content upon dilution, may explain the increased in-vitro release rate and extent.53
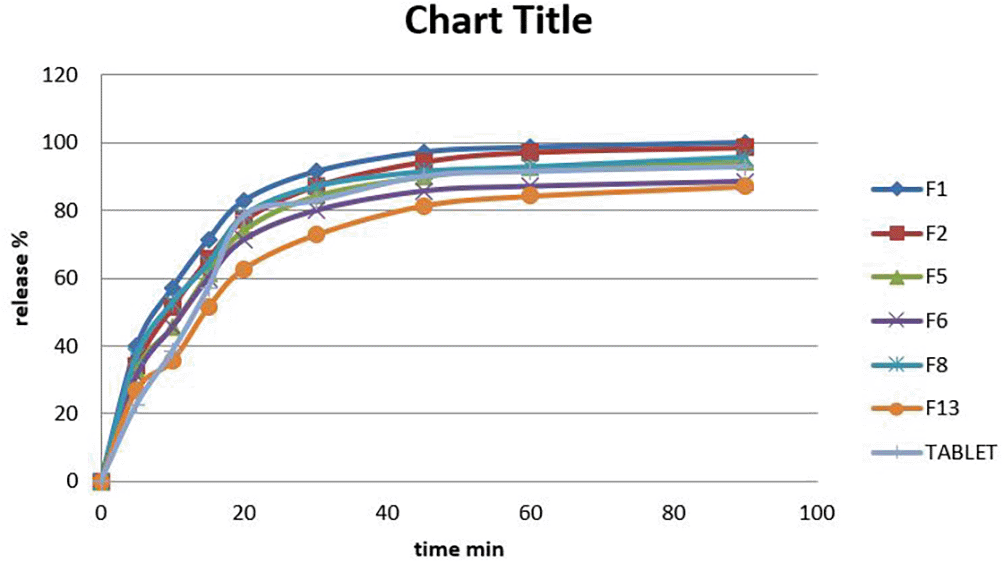
As compared the release from the formulation with constant oil% w/w and an increase in Smix, a clear decrease in norfloxacin releasing rate was seen because high Smix may cause drug sequestration inside micelles or oil droplets.29 It was discovered that increasing the co-surfactant yielded the highest dissolution rate. The reason for this is that co-surfactant works as a solubilizer, providing high thermodynamic activity of norfloxacin and therefore facilitating dissolution.47,54
When drug release was compared between optimum liquid SEDD formula of norfloxacin (F1) and norfloxacin commercial Tablet in ddsolver; the F1 formula showed a difference than tablet (f1 = 15.25, If (f1) ∈ [0-15], this means the reference formula is similar to tested formulations) higher release than the commercial Tablet as shown in Figure 3, and this is related to the ability of F1 to form spontaneous nano-emulsion.

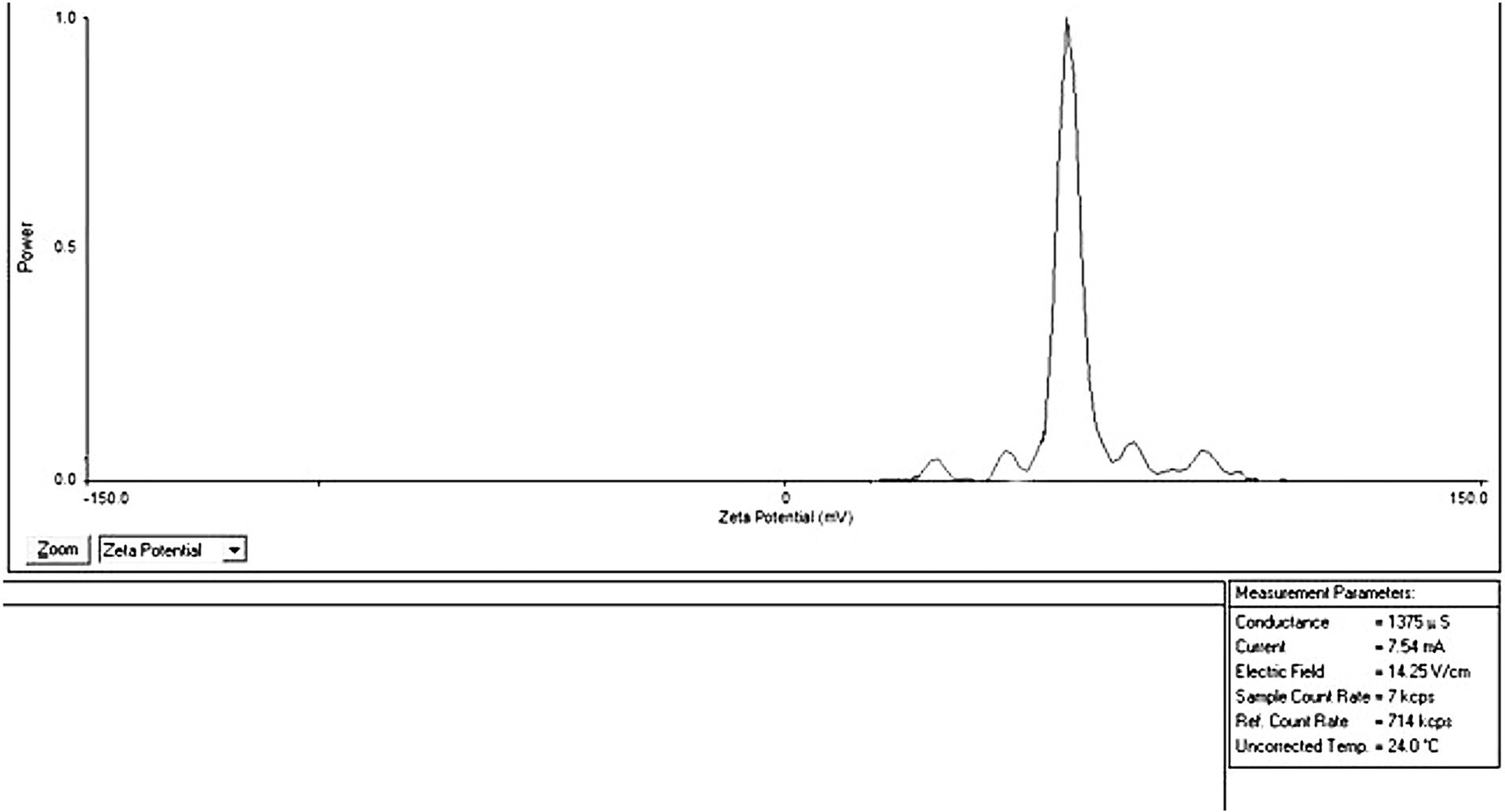
According to the thumb rule, the zeta potential is an essential predictor of the nanoparticle stability in dispersion medium when the result between (-5 mV to +5 mV) suggests quick agglomeration and the result between (-20 mV to +20 mV) gives short-term stability. Levels of zeta potential between (-30 mV) to (+30 mV) suggest moderate stability, while values in the range (-60 mV) to (+60 mV) indicate excellent stability in the formulation.55 The calculated zeta potential for the best liquid SEDD formula F1 was +60.78, as showen in Figure 4 indicating excellent stability.
An oral administration of spontaneous dosage form of norfloxacin that can produce in-situ nano-emulsion in the gastrointestinal tract providing a stable SEDDS that may increase drug absorption and bioavailability was successfully developed for the first time. Norfloxacin liquid dosage form is also required since a number of individuals, particularly pediatric and geriatric patients, have difficulties swallowing solid dosage forms.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
{kind=link}
Comments on this article Comments (0)