Keywords
Genetic, Malignancy, Aplastic anemia, Bone marrow depression, Prenatal testing
This article is included in the Datta Meghe Institute of Higher Education and Research collection.
Genetic, Malignancy, Aplastic anemia, Bone marrow depression, Prenatal testing
Swiss paediatrician Guido Fanconi was the first to discover Fanconi anaemia (FA) in 1927. He noticed that three boys had similar birth defects and died due to a condition mimicking pernicious anaemia. Later on, in 1960, motivated by Fanconi’s work, it was found that the disorder was due to chromosomal abnormalities and could cause bone marrow failure and malignancy. With recent advances in genetic studies, it is now known that FA is caused by changes in 22 genes. It is autosomal recessive but can also be autosomal dominant or X-linked recessive. According to the research, FA protein serves as a regulator of inflammatory cytokine and cell cycle via the p53/p21 pathway, as well as oxidative phosphorylation. Witnessing technological advancements that allow us to understand complicated human genetics further is genuinely awe-inspiring. Interference in any of these functions leads to the depletion of hematopoietic stem cells (HSCs), which leads to bone marrow failure and skeletal anomalies.1
Around the world, the incidence of Fanconi anemia is 1 in 160,000. It is observed that FA is presented among people of Ashkenazi Jewish descent, the Roma population of Spain, and Black South Africans.2 In India, Fanconi anemia has an incidence of 1 per 3,60,000 live births.3
Clinical finding in FA includes pancytopenia, hyperpigmentation or hypopigmentation, skeletal anomalies, small stature or growth retardation, endocrine abnormalities, anal atresia, deafness, malignancy of the head and neck, and anorectal and familial occurrence.4
Diagnosis of FA can be made on clinical findings and laboratory examination. Recommended prenatal testing like chorionic villus sampling and amniocentesis is advised within ten and fifteen weeks of gestation. If genetic variants are known, then targeted variant analysis can be done, whereas if familial variants are not known, then chromosome breakage testing is recommended. Androgen therapy and hematopoietic stem cell transplantation (HSCT) are treatments recommended for FA. Treatment with gene therapy as part of a clinical trial in the field of medicine holds immense promise for patients with various genetic disorders including FA. The progress being made gives hope for the future.5
Follow-up of all diagnosed FA patients is essential throughout their lifetime. This includes monitoring haemoglobin levels every 3–4 months. Treatment should be started when the haemoglobin level decreases below 8 g/dL. To maintain good health after diagnosis, it’s recommended to have a complete blood count (CBC) every six months and bone marrow studies annually, especially if the patient has a normal complete blood count (CBC) at the time of diagnosis. This approach will help identify any potential health issues and ensure that the patient receives the appropriate treatment if necessary. If a patient is diagnosed with cytopenia, it is recommended that they undergo CBC every three months and bone marrow studies every six months. Blood glucose levels should be monitored every two weeks as FA has changes in insulin secretion because of abnormalities in glucose metabolism. Magnetic resonance imaging (MRI) is studied to rule out malignancy every six months.
It’s crucial to have a meticulous treatment plan with supportive care from a multidisciplinary healthcare team. This may involve working with an ear nose throat (ENT) surgeon for audiological concerns, a dermatologist and endocrine specialist for hypogonadism and thyroid dysfunction, regular dental check-ups for oral health, and consultations with an ophthalmologist, dietician, and nutritionist. By taking a proactive approach and collaborating with your healthcare team, potential health complications can be identified and improve overall wellbeing.
Our aim of the study is to increase awareness among physicians about the presence of such rare diseases. Well-time diagnosis will permit appropriate treatment to start before complications develop. Also, it will allow parental decision–making about prenatal diagnosis in their future pregnancy.
An 8-year-old male child of Indo-Aryan ethnicity, came to the tertiary centre in the paediatric ward with complaints of an increase in the rate of respiration, pallor, fever, and cough for 10 days. He had a history of similar complaints on and off since birth. Antenatal or postnatal history was uneventful. Psycho-social history was not significant. There was no history of congenital anomalies in the family. Personal and developmental history was normal for the patient’s age. Anthropometry examination showed height for age and head circumference around the 50th percentile. On the physical examination, the patient has an absent right thumb and a rudimentary small left thumb (Figure 1).

The patient had pallor, no icterus, and no significant lymphadenopathy. The right kidney was not seen in ultrasonography. Pancytopenia with anisocytosis was seen in the peripheral examination. The normal range of haemoglobin in this age group is 11.9 to 15 g/dl. The patient’s haemoglobin was 6 gm/dl. The platelet count was 17,700/μL, which gradually improved on receiving packed red blood cell transfusion (PRBC) and platelet concentrates (PC) transfusions. The normal range of antinuclear antibodies (ANC) is 1:160 to 1:180. In the present patient, ANC was 1:20 which was on the lower side. Blood examination for vitamin B12 and folic acid values were normal. Bone marrow analysis showed signs of hypocellular bone marrow, with reductions in all cell lines (erythroid, myeloid, megakaryocytic) which is indicative of aplastic anemia. There was no sign of fibrosis, granuloma, or metastasis on the magnetic resonance imaging (MRI) or computed tomography (CT) scan. Echocardiography was normal. The thyroid level was within the normal limit. Liver function test and kidney function test were within the normal ranges. Normal blood sugar level at this age is 90 to 180 mg/dl; the patient’s blood sugar was 80mg/dl. Oxygen saturation was 88% at time of admission so oxygen administration was initiated. X-ray of the chest showed bilateral infiltrates suggestive of pneumonia, for which the patient was started on intravenous third generation antibiotics, ceftriaxone 500 mg given every 8 hours for 5 days, later patient was shifted on tablet cefixime 100 mg two times a day for next 5 days. X-Ray of the right arm showed that the radius bone was absent along with bowing of the ulna (Figure 2).
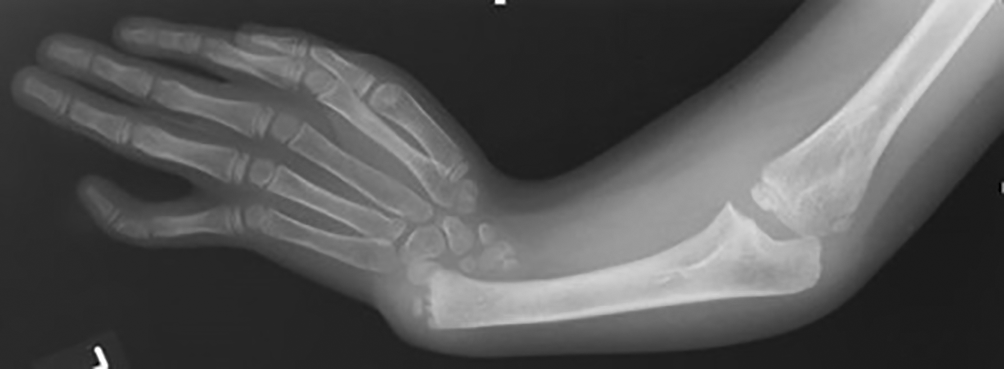
Because of the absence of a thumb and radius bone, absent right kidney on ultrasonography, and aplastic anaemia on bone marrow examination, we thought of going for a genetic test. A chromosomal breakage study for Fanconi anaemia was done, which showed metaphase karyotyping; GTG banding (G-bands by trypsin using Giemsa) showed karyotype-46XY, abnormal chromosomal breakage syndrome with significant structural aberrations in most of the metaphases, which confirmed the final diagnosis for FA.
Other examinations correlated with FA were done, which included an ear examination that did not show any sign of deafness. Test for growth hormones, insulin, and lipid profile were in the normal range. The thyroid function test and glucose tolerance test were in the normal range. Referral to the orthopaedic department helped the patient obtain the functional and cosmetic outcomes needed for their thumb abnormalities.
The risk factor associated with FA was explained to parents, follow-up sessions were scheduled, and the patient was discharged after 15 days of admission. Prophylaxis iron therapy at a dose of 3mg/kg, 2.5 ml once a day through oral route was started and will be continued for three months.
During the follow-up, we planned for appropriate follow-up care for the patient to ensure that they receive the necessary treatment and support. The patient was advised to test for haemoglobin and CBC to be done every three months and bone marrow studies every year. Blood glucose levels should be monitored every two weeks as FA has changes in insulin secretion because of abnormalities in glucose metabolism. Magnetic resonance imaging (MRI) will be done to rule out malignancy every six months. Every six months, the patient was advised to have an appointment for consultation with ENT surgeon, dermatologist, endocrinologist, ophthalmologist to rule out any malignancy.
The prognosis and outcome were good in the present study patient as he did not show any tumour or bone marrow failure on follow-up.
FA is a rare inherited disease associated with various abnormalities like absent limbs, absent kidneys, anaemia, urogenital anomalies, and bone marrow aplasia. It is caused by defects or mutations in genes. The protein that is synthesized by this gene helps to recognise and regenerate damaged DNA. In FA, because of defects in genes, the DNA regeneration process diminishes, and new stem cells are not produced, leading to aplastic anaemia.6
FA is seen in the first decades of life. Males are more affected than females. Ghaida Bakoar Alahmadi et al. in their study, showed that the mean age of presentation of FA was about 7–8 years and it had a male predominance, which was similar to the present study.7
The benchmark for diagnosing FA is on chromosomal breakage study for metaphase karyotyping; GTG banding, which shows abnormal chromosomal breakage syndrome with significant structural aberrations in most of the metaphases. Bushra Anam Ali et al. studied Fanconi anaemia on chromosomal breakage and found abnormal chromosomal breakage syndrome with significant structural aberrations in most of the metaphases, which was seen in the present study.8
Parental consanguinity was common in a study by Heather A et al. that highlighted the importance of community and youth education and the importance of pre-marital counselling. This was opposite to the present case report as parenteral consanguinity was not seen, nor was pre-marital counselling done.9
Of the many physical abnormalities, hearing loss is commonly observed with FA. In their research, M D Tischkowitz et al. showed that conductive deafness is common and may or may not be associated with external ear malformations. This was not seen in the present study.10
Congenital anomalies in the renal system are associated with FA. Vijaya Sathyanarayana et al. showed that renal abnormalities are commonly seen in FA. In the present study, absence of a kidney was seen on the right side, which was similar to the study done by Vijaya Sathyanarayana et al.11
Short stature, obesity, glucose intolerance, and hypothyroidism are some of the endocrine abnormalities associated with FA. In their research, Neelam Giri et al. showed that affected males with FA have a high incidence of endocrine abnormalities, which was not seen in the present study.12
Malignancy is associated with FA. Philip S Rosenberg et al. showed a higher incidence of malignancy in FA, like leukaemia and solid tumours, which were not seen in the present case report.13
A Butturini et al. presented data suggesting that FA subjects have an impairment of haematopoiesis that leads to hypoplastic anaemia and bone marrow failure during childhood; this was similar to the present study in which the child showed signs of bone marrow failure on admission.14
Thumb anomaly and the absence of a radius on radiological examination was seen in the present case, which is rare in FA. Based on the review conducted by B.P. Alter, it has been observed that paediatric patients who have congenital anomalies of the upper limb are often associated with genetic disorders of the bone marrow. These disorders can lead to bone marrow failure, such as FA and others similar to the present case.15
It is important to note that prenatal testing and monitoring for malignancy and bone marrow examination can increase the chances of survival for paediatric patients with congenital anomalies of the upper limb. However, it is also important to consider the limitations of the study, such as the potential for patients to become tired of visiting the hospital for follow-up appointments and a need for a larger sample size for effective follow-up. Proper management and care for these patients is crucial, including consideration of genetic disorders that may lead to bone marrow failure.
As the patient is a child his perspective is difficult to judge, but the parents were very anxious and nervous as long-term follow-up brings uncertainty. What will happen in the follow-up was the question commonly asked. Parents need to be counseled by educated and empathetic paediatricians who can provide comfort and support during what can be a very challenging time. While prenatal testing and monitoring, as well as bone marrow examinations, can increase the chances of survival, it’s important to be aware of potential limitations. Follow-up appointments are an absolute must, and a larger sample size is needed for effective follow-up. Additionally, genetic disorders that may lead to bone marrow failure should be considered in the overall care plan for these patients. This is not something that can be overlooked or ignored.
Fanconi anaemia is associated with aplastic anaemia, physical anomalies, bone marrow failure, and malignancy. Since this is rare, physicians should be aware of this condition so that other associated complications can be prevented and treated as well. Parents should be well-informed of the challenges that may arise when caring for a child with congenital anomalies of the upper limb. Correct management and care are extremely important, which is why parents should seek assistance from knowledgeable and sympathetic paediatricians who can provide much-needed support during this time. Regular follow-up appointments with a multidisciplinary team are essential, as is close surveillance and treatment. It’s important for parents who have had children diagnosed with FA to be cautious and plan ahead when considering subsequent pregnancies. Above all, it’s imperative to be mindful of potential limitations and genetic disorders that may have an impact on a child’s care plan.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)