Keywords
Alzheimer's disease, BACE1, Molecular docking, Molecular dynamics, Natural compounds, Hinokiflavone
Alzheimer's disease, BACE1, Molecular docking, Molecular dynamics, Natural compounds, Hinokiflavone
Alzheimer’s disease (AD) is a progressive neurological disorder that declines in cognitive function and is usually recognized as a major health hazard that mostly affects people older than 60 years old (Husna Ibrahim et al., 2020). It occurs due to a gradual and slow neurodegenerative process, leading to age-related neuronal cell death. The disease is characterized by the accumulation of amyloid plaques, the tangling of neurofibril tissue, and synaptic dysfunction. It manifests in various stages, gradually affecting everyday functions and leading to symptoms like apathy, depression, communication difficulties, disorientation, impaired judgment, swallowing and walking problems, and behavioral changes (Abubakar et al., 2022).
Regrettably, there is currently no known cure for the disease. Nowadays, researchers are dedicated to comprehending the pathology of AD through various mechanisms, such as abnormal tau protein metabolism, β-amyloid, inflammatory response, and cholinergic and free radical damage, aiming to develop effective treatments that can stop or slowing the progression of AD (Breijyeh & Karaman, 2020). To prevent the death of neurons, one potential approach involves inhibiting beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), which can help prevent the buildup of beta-amyloid (Aβ). This strategy aims to address one of the underlying mechanisms associated with AD pathology (Yan & Vassar, 2014).
BACE1, an enzyme discovered in 1999, plays a crucial role in the early stages of AD (Yan & Vassar, 2014). BACE1 is an enzyme responsible for breaking down the Amyloid Protein Precursor (APP) and releasing APP fragments, which lead to the formation of Aβ on nerve cell membranes, which is a key component of the plaques associated with AD. It is found at higher levels in the brains and body fluids of individuals with AD, suggesting its involvement in the disease. Therefore, targeting BACE1 activity with drugs may hold potential for therapeutic interventions aimed at reducing Aβ accumulation and slowing down the progression of AD (Das & Yan, 2017).
In the drug development process, docking plays an important role in drug design. Molecular docking has been used to search low binding free energy and also the ligand conformations at the protein-binding sites. In addition, docking also provided useful information for predicting the orientation of drug candidate binding to the protein target (Pinzi & Rastelli, 2019). The focus of our study was on conducting a multistage virtual screening of 400 natural compounds extracted from natural plants. These compounds were carefully chosen based on a thorough review of the existing literature, specifically focusing on selecting molecules with known anticancer activity. The objective was to identify natural compounds that exhibit high affinity towards inhibiting BACE1 activity while having low toxicity.
In this research, we selected a database of 400 BACE1 inhibitor compounds (Elkamili et al., 2023). We used the PubChem library to retrieve all the ligand compounds, in SDF format. These compound structures were then imported into OpenBabel for conversion into PDB format (O’Boyle et al., 2011). The compounds were further prepared using AutoDockTools, during which Gasteiger charges were applied to them and saved in PDBQT format.
The target protein, BACE1 (PDB code: 3K5D), was prepared before commencing the docking process. The three-dimensional crystal structure of the protein was obtained from the PDB database in PDB format. In AutoDockTools, the structure was processed by removing water molecules and heteroatoms, and then Kollman charges and polar hydrogens were added and the structure of the protein was saved in PDBQT format.
Recognizing the suitable active site for binding the ligand molecules is the most crucial aspect in designing a drug via computational docking. AutoDock Vina (RRID:SCR_011958) was employed for molecular docking at exhaustiveness level 8 to predict the potential interactions between the ligand compounds and the protein BACE1. The grid box of the protein structure was adjusted to fit the active site that is predicted by CB-Dock2 to predict the binding pocket of the BACE1 target (Liu et al., 2022). The spacing was set to 13 Å, 8 Å, and 70 Å points along the x, y, and z-axe, with the lattice size set to 27 Å × 27 Å × 27 Å at the center. A total of 400 compounds were screened and docked against the target protein. Docking affinity was used to generate nine poses for each protein-ligand complex.
In our study, we employed Lipinski’s rule of fives using SwissADME as a guideline for evaluating the drug-likeness and potential oral bioavailability of the selected compounds with best affinities (Daina et al., 2017). By assessing factors such as molecular weight, lipophilicity, hydrogen bond donors and acceptors, and the number of rotatable bonds, Lipinski’s rule helps in identifying compounds that possess favorable physicochemical properties for drug development. In our screening process, we applied these criteria to filter out compounds that were more likely to exhibit optimal pharmacological activity, thus narrowing down our selection to the most promising ligands for further analysis and potential therapeutic applications.
To predict the potential toxicity of the compounds, the ProTox-II web server was utilized. ProTox-II is a reliable online tool specifically designed for predicting the toxicity of chemicals (Banerjee et al., 2018). By inputting the SMILES of the compounds into the server, we obtained valuable toxicity prediction results. This step was crucial in identifying and excluding potentially toxic molecules, ensuring that only safe compounds were included in the subsequent analyses and minimizing potential harm based on several types of toxicity that were evaluated including hepatotoxicity, carcinogenicity, cytotoxicity, mutagenicity, immunotoxicity, and DL-50.
Biova Discovery Studio, a comprehensive molecular modeling and simulation platform, offered a wide range of features and modules that facilitated the investigation of complex molecular systems through the visualization of interactions of post-docking complexes, facilitating a deeper understanding of the interactions between the compounds and their respective targets.
The MD simulations were conducted for 100 nanoseconds to ensure the stability of interactions between hinokiflavone and the BACE1 target using Schrodinger LLC Desmond software (RRID:SCR_014575). Before the simulations, the ligand-receptor complex was subjected to preprocessing using Maestro’s Protein Preparation Wizard, which involved optimization, and minimization. The system was then constructed using the System Builder tool (a module of Protein Preparation Wizard). For the simulations, the TIP3P solvent model was employed in an orthorhombic box, maintaining a temperature of 300 K, pressure of 1 atm, and utilizing the OPLS force field. To simulate physiological conditions, counter ions, and 0.15 M sodium chloride were added to neutralize the models. Before the actual simulation, the models were equilibrated, and trajectories were saved at intervals of 100 ps for further analysis and inspection.
We used the Cb-Dock2 website to predict the binding pocket of the BACE1 target. Figure 1 displays the key residues of the potential binding site on the protein (Elkamili et al., 2023). All of these residues are located in chain C which include: ASP32, GLY34, PRO70, TYR71, THR72, GLN73, GLY74, LYS107, PHE108, PHE109, ILE110, TRP115, ILE118, TYR198, LYS224, ILE226, ASP228, GLY230, THR231, THR232, ARG235, THR329, GLY330, THR331 and VAL332. These residues have been mentioned in several studies as part of the active site of BACE1 (Wu et al., 2016; Xu et al., 2012; Hernández-Rodríguez et al., 2016; Ellis & Shen, 2015).
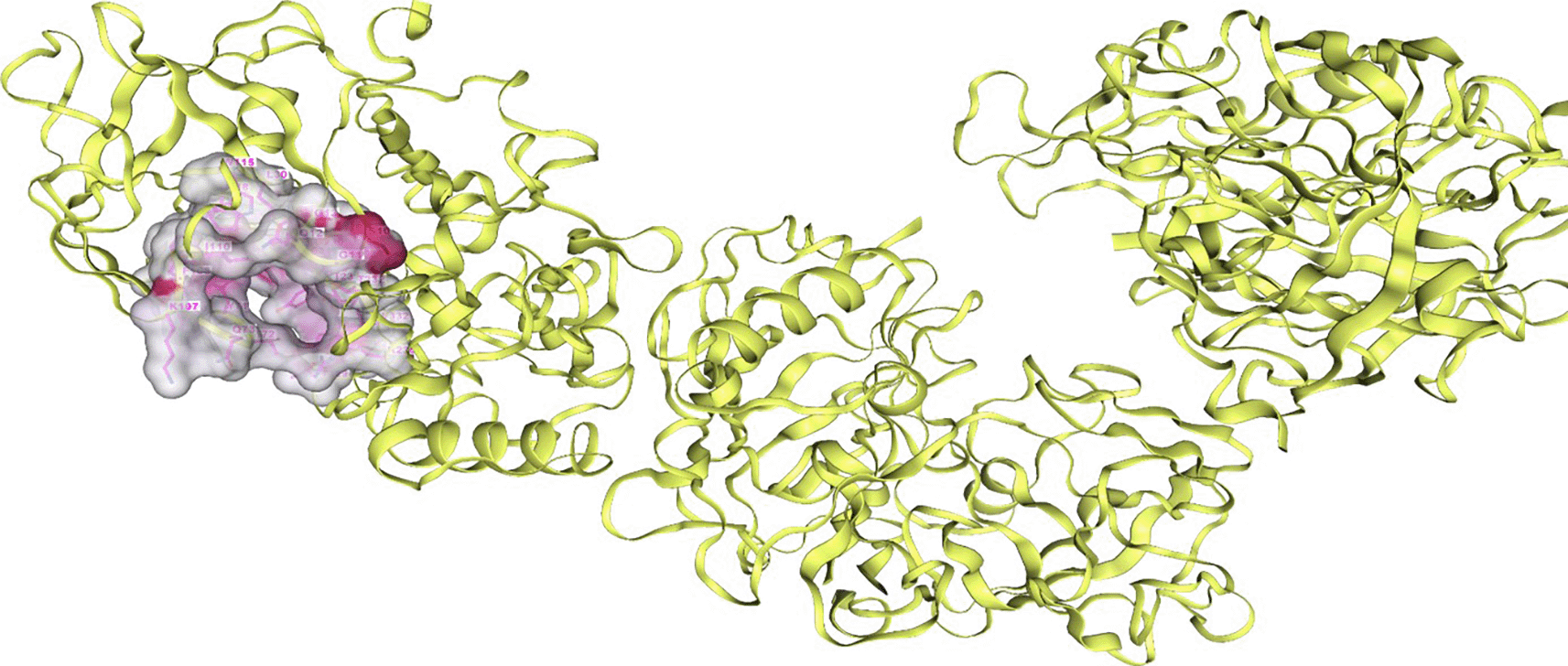
Key residues represented in pink in the binding pocket and the surfaces of the binding pocket in BACE1 are presented in grey. BACE1, Beta-site amyloid precursor protein cleaving enzyme 1.
A total of 400 compounds were screened against the protein BACE1 using AutoDock Vina to identify potential binding interactions and determine the optimal binding affinity. The docking results yielded positive outcomes, demonstrating a strong binding affinity for the screened compounds. The lowest binding affinity, at -10.8 kcal/mol, was observed with hinokiflavone. On the other hand, the highest binding affinity reached -4.2 kcal/mol with epigoitrin. For further analysis, we focused on compounds with scores equal to or below -9 kcal/mol to identify molecules with significant potential for therapeutic efficacy, reliability, and favorable interactions. A list of the selected compounds with the best binding affinity, obtained after the analysis of the docking results can be found in Table 1. We used these molecules, which demonstrated binding affinity scores equal to or lower than -9 kcal/mol, to apply the Lipinski’s Rule of Five parameters and select potential drug candidates.
After conducting the molecular docking analysis, we examined the results to identify the top 59 molecules exhibiting the strongest binding affinity with the target protein. From this initial set, we further refined our selection and focused on 41 compounds that displayed exceptional properties, showing great potential for further development as potential drug candidates. These 41 compounds successfully passed the Lipinski’s Rule of Five test, validating their suitability for further investigations in our future drug development efforts. These compounds are presented in Table 2.
The second part of the filtration implicate the utilization of ProTox-II as a filtering tool for compound analysis based on hepatotoxicity, carcinogenicity, cytotoxicity, mutagenicity, immunotoxicity and LD50 (lethal dose) characteristics proved to be highly effective in isolating non-toxic and safe molecules. The application of ProTox-II played a crucial role in the evaluation of the compounds, allowing for a comprehensive assessment of their safety profiles. This step was essential to ensure that only compounds with favorable safety characteristics were selected for further investigation. Out of the initial pool, only hinokiflavone which was identified as inactive in all targets, and the only molecule that exhibits slight toxicity, with a LD50 (lethal dose) closer to 5000, these results are presented in Table 3. Therefore, hinokiflavone is considered a safe natural compound emerging as a potential candidate for molecular dynamics simulations to validate its stability in complex with BACE1 under physiological conditions.
The visualization of the 2D interactions of the BACE1-hinokiflavone complex is performed using Discovery Studio enabling the prediction of protein residues with which the ligand interacts to validate if the candidate compound hinokiflavone interacts with the active site residues of the BACE1 target. Figure 2 showed that hinokiflavone strongly interacted with most of the active site residues of BACE1, indicating that our results perfectly corroborate with the active site residues of BACE1, justifying their anti-Alzheimer’s potency. In summary, hinokiflavone binds to BACE1 via van der Waals interactions with ASP32, TYR71, GLN73, GLY74, PHE108, PHE109, TRP115, ILE118, LYS224, GLY230, THR231, THR232, GLY330, and VAL332. It forms hydrogen bonds with LYS107, TYR198, and THR329, pi-alkyl interactions with ILE110 and ILE226, pi-anion interaction with ASP228, and a pi-donor hydrogen bond with THR72. Furthermore, molecular dynamics simulations allow for the exploration of the dynamic behavior and stability of the ligand-receptor complexes over time.
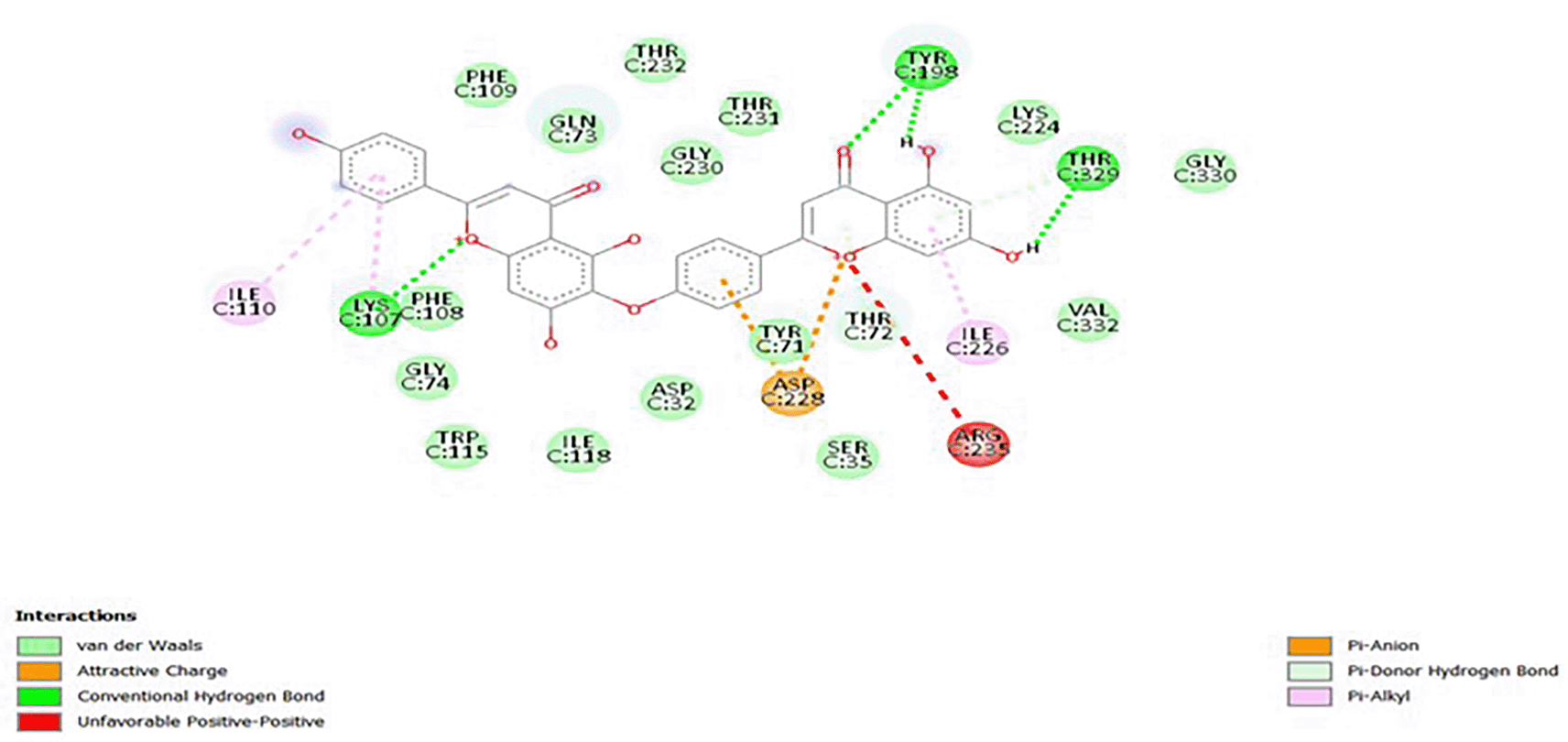
BACE1, Beta-site amyloid precursor protein cleaving enzyme 1.
The dynamic simulation of proteins plays a crucial role in understanding their behavior and interactions. Unlike static molecular docking, dynamic simulations consider the flexibility and conformational changes of proteins over time. This allows for a more comprehensive and accurate analysis of protein-ligand interactions. In this study, we employed dynamic simulations to investigate the dynamic behavior of the BACE1-Hinokiflavone complex, providing valuable insights into its physiological relevance and enhancing our understanding of its functional mechanisms. To capture the dynamic nature of the BACE1 protein, we conducted 100 nanosecond molecular dynamics simulations based on the rigid crystal structure of the protein. By doing so, we gained valuable insights into the dynamic behavior of the BACE1-Hinokiflavone complex, enhancing the physiological relevance of our findings. These simulations allowed us to observe conformational changes and interactions that are crucial for understanding the functional mechanisms of the complex. The parameters explored for analysis included root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein secondary structure, and protein-ligand contact analysis.
The calculation of RMSD allows us to determine the stability and balance of the complex throughout the 100 ns simulation. A low RMSD value indicates a stable complex, while higher values suggest instability. Furthermore, fluctuations within a broad range indicate significant conformational changes in the protein during the simulation, although fluctuations of approximately 1-3 Å are perfectly acceptable. In the simulation (Figure 3), the left Y-axis provides an overview of the protein’s RMSD evolution, while the ligand’s RMSD graph (right Y-axis) indicates the ligand’s stability within the protein and its binding pocket. The Root Mean Square Deviation (RMSD) of Hinokiflavone bound to the BACE1 protein (Figure 3) remained stable without significant structural deviations after 40 ns towards the end of the simulation, indicating a balanced conformation. This allows for an extended simulation for rigorous analysis.
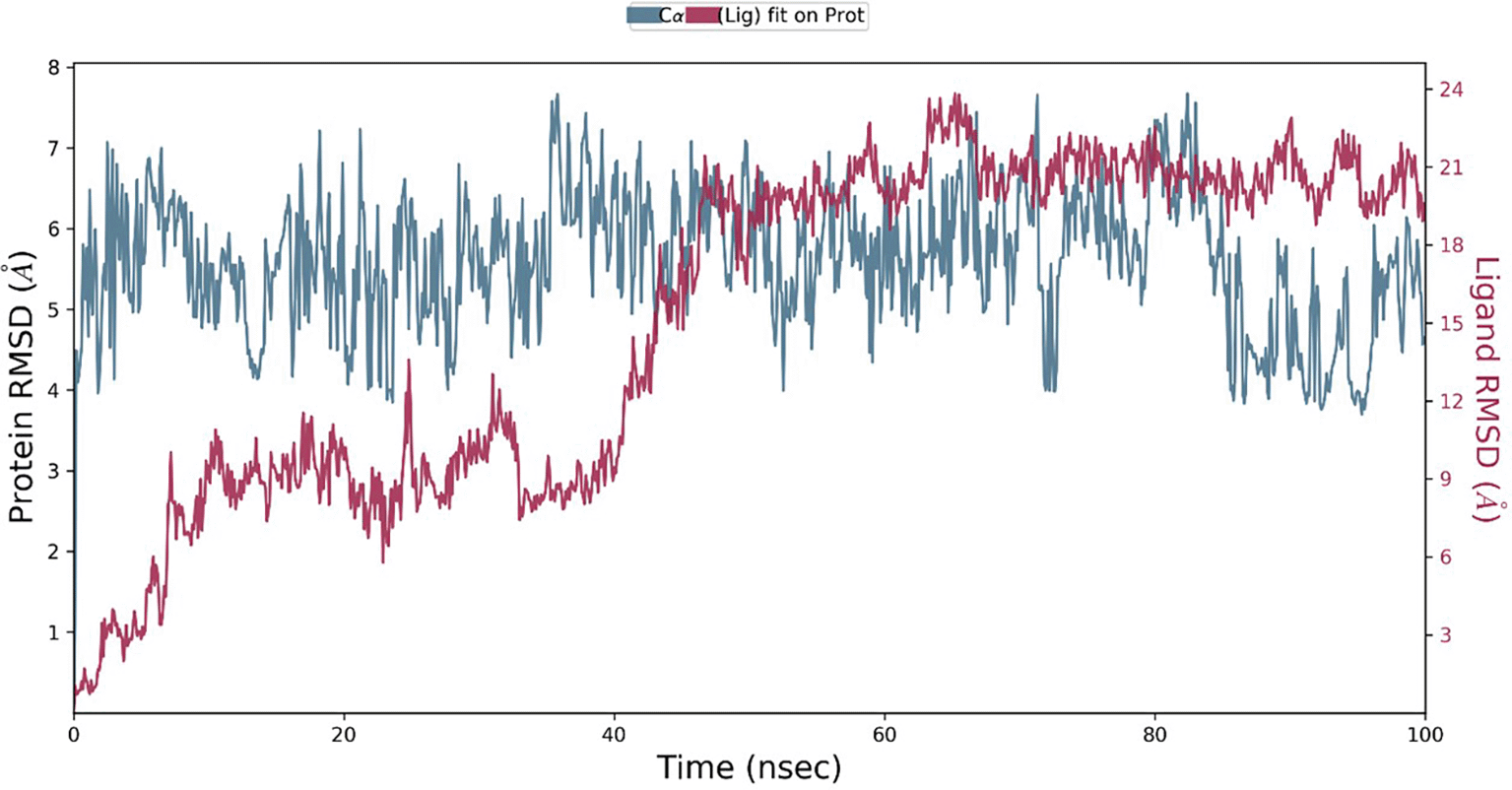
RMSD, Root Mean Square Deviation; BACE1, Beta-site amyloid precursor protein cleaving enzyme 1.
RMSF is a measure of particle deviation within the protein, revealing the flexibility and rigidity of each amino acid throughout the simulation. Lower RMSF values indicate good system stability, and vice versa. Generally, the terminal regions (N- and C-terminal) exhibit more fluctuations compared to other parts of the protein. However, secondary structure elements such as alpha helices and beta sheets are typically more rigid and experience less fluctuation than loop regions. The RMSF plot (Figure 4) remained stable throughout the simulation, with some fluctuations stabilizing in the ligand-binding regions (indicated by green-colored vertical bars). This indicates that Hinokiflavone contributes to the stability of the BACE1 protein.
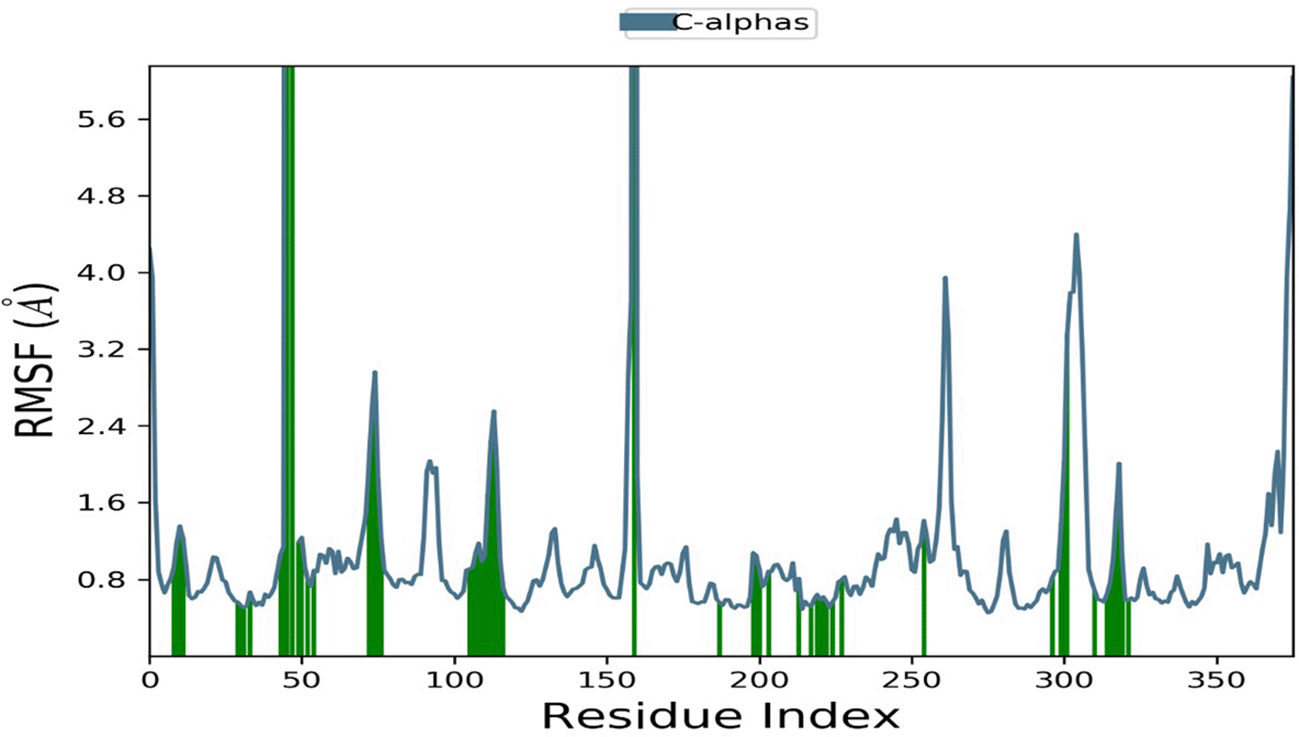
The vertical green lines represent the amino acid residue of BACE1 making contact with ligand. RMSF, Root Means Square Fluctuation; BACE1, Beta-site amyloid precursor protein cleaving enzyme 1.
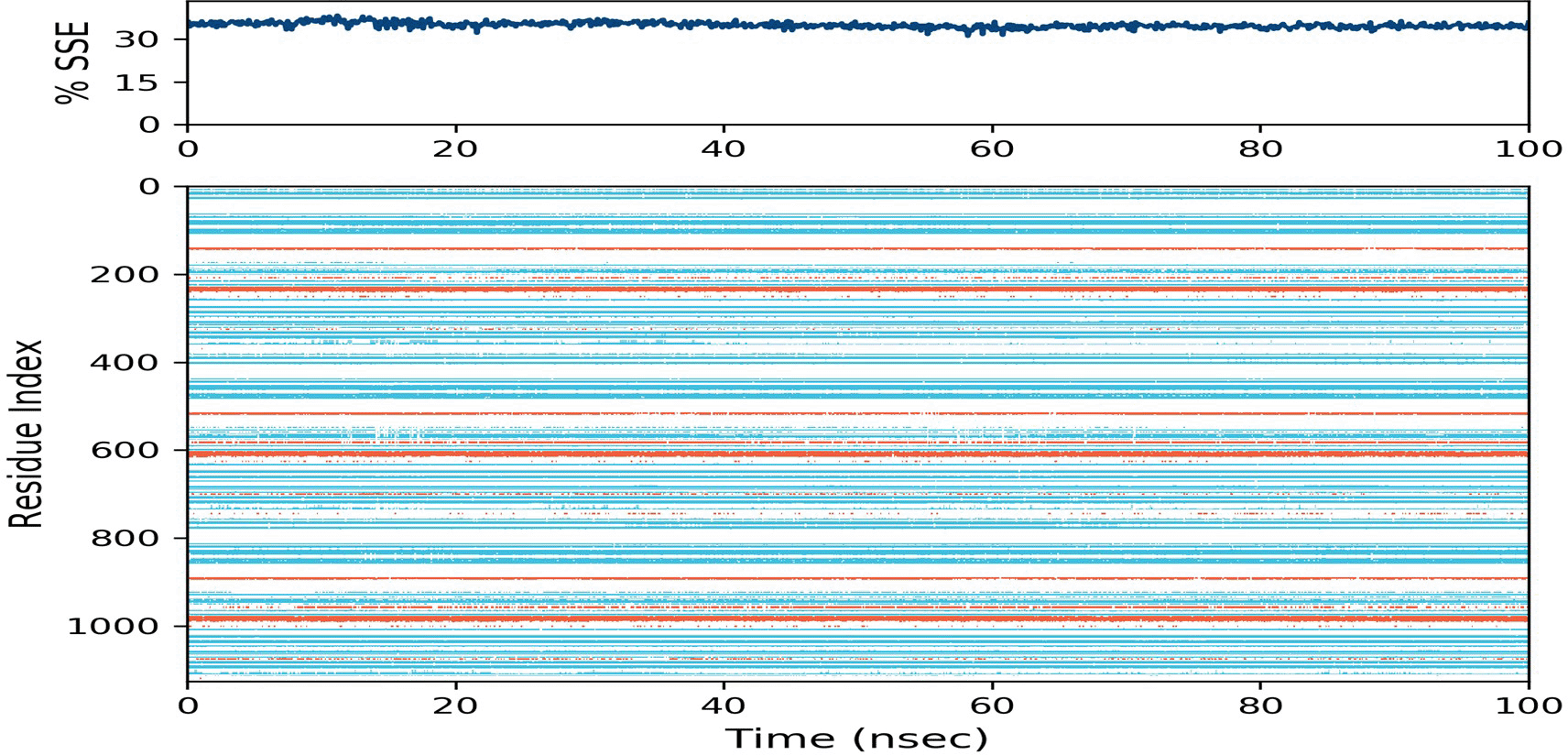
Protein secondary structure elements (SSE) like alpha-helices and beta-strands are monitored throughout the simulation. The plot at the top summarizes the SSE composition of the protein over the course of the simulation, and the plot at the bottom monitors each residue and its SSE assignment over time, with variations represented in orange and blue corresponding to alpha helices and beta sheets, respectively (Figure 5). Figure 5 showed that the percentage of protein secondary structure elements in BACE1 was approximately 37% and remained constant throughout the simulation, indicating the stability of the overall structure of BACE1 during binding with hinokiflavone and thus the stability of the complex.
The interaction between the target protein and the ligand was monitored during the simulation to highlight the residues involved in this interaction. These interactions were categorized into four types: hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges. The interaction diagram between BACE1 and hinokiflavone (Figure 6) showed that hinokiflavone formed bonds with all the residues in the active site. Among them, GLN73, GLY230, and THR232 were maintained for more than 60% of the simulation time, and all three interactions were hydrogen bonds mediated by water.
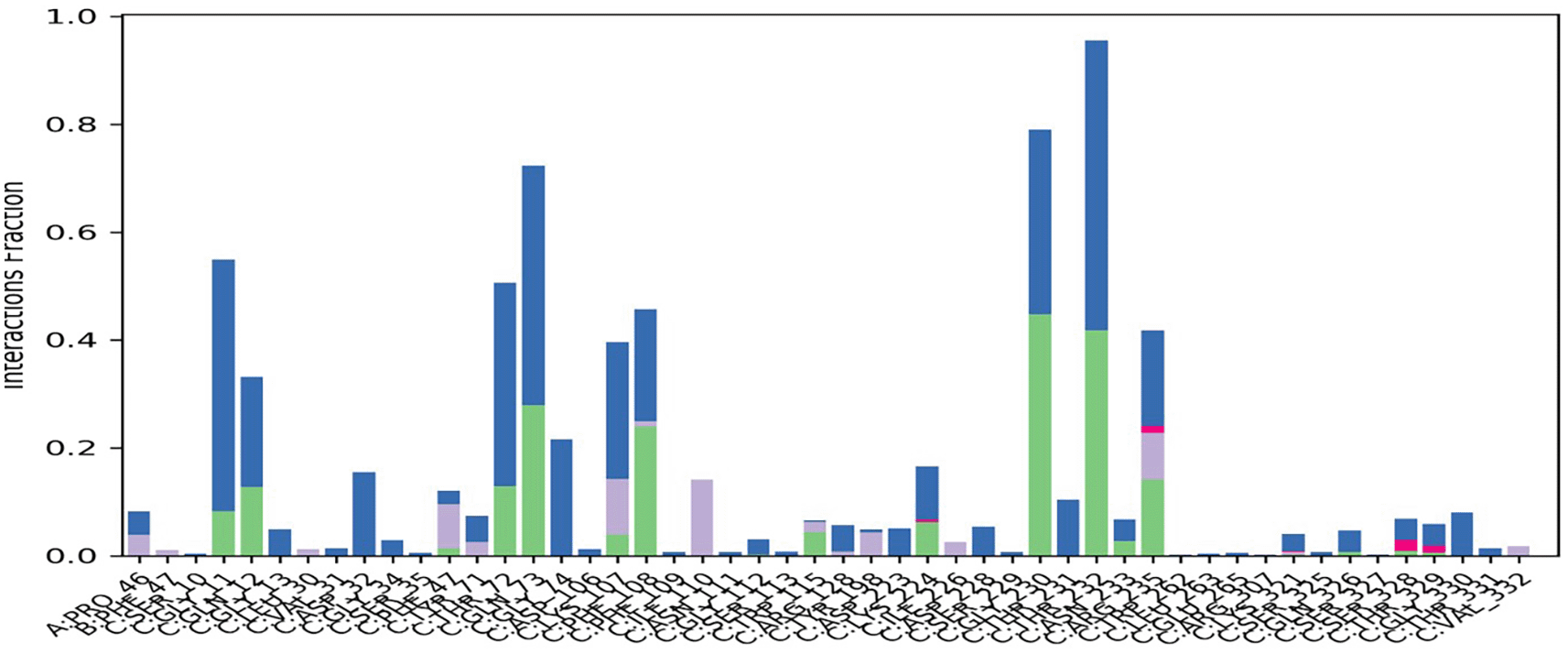
Different color signifies different interactions and contacts formed between the amino acids and the ligand atoms. BACE1, Beta-site amyloid precursor protein cleaving enzyme 1.
In conclusion, the findings of this study hold significant importance in the field of AD research. By targeting the BACE1 protein, which plays a crucial role in the production of amyloid-beta peptides, the progression of AD can potentially be decelerated. The use of computational methodologies, including molecular docking and molecular dynamics simulations, allowed for the identification of novel inhibitors against BACE1. Among the natural compounds reviewed, hinokiflavone emerged as a highly promising candidate. It exhibited strong binding affinity and favorable binding free energy values, indicating its potential effectiveness as a BACE1 inhibitor. Moreover, hinokiflavone successfully met the criteria of Lipinski’s rule of five, suggesting its drug-like properties and potential for development as a therapeutic agent. The molecular docking results were further supported by molecular dynamics simulations, which demonstrated the stability of hinokiflavone within the binding site of BACE1 over a 100 ns trajectory. This stability indicates the potential of hinokiflavone to effectively interact with the target protein and inhibit its activity. Overall, the discovery of hinokiflavone as a potential BACE1 inhibitor represents a significant step forward in the search for novel treatments for AD. Further research and validation studies are warranted to fully understand the therapeutic potential of hinokiflavone and its role in mitigating the pathology associated with AD.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)