Keywords
next generation sequencing, illumina, nanopore, microbes, cultured species
This article is included in the Nanopore Analysis gateway.
next generation sequencing, illumina, nanopore, microbes, cultured species
In the past decade, next-generation sequencing (NGS) technology has revolutionized genomics research, enabling the analysis of DNA and RNA at an unprecedented scale and speed. The potential applications of NGS technology in diverse fields, such as medicine, agriculture, and environmental science, have fueled the development of this technology and its use across the globe. NGS has a wide range of applications in various fields of research, and is increasingly being used to advance knowledge and understanding of genetics, biology, and related fields, including genomics, transcriptomics, epigenomics, metagenomics, and proteomics (Satam et al., 2023). Next generation sequencing can be used to sequence the entire genome of an organism, providing information about its genetic makeup and variation, sequence the transcripts such as messenger RNA of an organism, providing information about its gene expression patterns, sequence epigenetic modifications such as DNA methylation and histone modification, providing information about how gene expression is regulated, sequence the DNA of a microbial community in an environmental sample, providing information about the diversity and function of the community, and sequence peptides or proteins, providing information about protein structure and function. The versatility of next-generation sequencing makes it a valuable tool for research and discovery in many different areas of science. Some of the fields involved in the application of next generation sequencing are cancer research, forensic science, agriculture and plant science and aquaculture. NGS technology has seen an increasing level of interest and investment (Behjati & Tarpey, 2013). The next-generation technology application has a unique and diverse ecosystem, including a vast array of flora and fauna, as well as a rich human diversity with more than 600 ethnic groups and a population of over 600 million people. There is a great potential for NGS technology to contribute to the region’s development in various fields, including biomedical research, agriculture, and environmental monitoring. Therefore, this systematic review aims to provide a comprehensive overview of the application of NGS technology with a focus on the fields of genomics, transcriptomics, epigenomics, metagenomics, and proteomics (Goodwin et al., 2016; Barba et al., 2014).
In recent years, there has been a growing trend towards using NGS in clinical applications. This includes the use of NGS for disease diagnosis, identification of genetic mutations, and personalized medicine. The availability of affordable NGS platforms and the development of robust bioinformatics tools have contributed to the growth of NGS in clinical settings (Qin, 2019). NGS is increasingly being adopted in agricultural research. NGS is being used to improve crop breeding programs, study plant genetics, and understand plant-pathogen interactions. The technology is also being used to study the microbiome of crops and soil, which can help improve crop yields and reduce the use of pesticides (Sharma et al., 2018). The use of metagenomics, which involves the sequencing of all the genetic material in an environmental sample, is emerging as a powerful tool. Metagenomics is being used to study the microbiome of soil, water, and gut microbiome, as well as to study microbial diversity in ecosystems. There is a growing trend towards the development of NGS-based diagnostics. This includes the development of tests for infectious diseases, cancer, and genetic disorders. The use of NGS-based diagnostics can provide more accurate and rapid diagnosis, leading to better patient outcomes. There is an increasing emphasis on collaboration and networking among researchers, with the aim of advancing NGS research in the region. Collaborative efforts are being made to share resources, exchange knowledge and expertise, and develop common research agendas. The trends and pattern of application of NGS driven by advances in technology, greater availability of resources, and increased collaboration among researchers in the region (Pereira et al., 2020; Børsting, & Morling, 2015; Liu et al., 2019).
The challenges and limitations discussed below highlight the need for more investment in infrastructure, expertise, and ethical and regulatory frameworks to support the use of NGS technology. By addressing these challenges, researchers can unlock the full potential of NGS to advance knowledge and understanding of genetics, biology, and related fields in the region. The high cost of NGS equipment, reagents, and data analysis software can be a major barrier to its use, particularly for researchers in low- and middle-income countries. To address this, some researchers have advocated for more affordable NGS technologies and have explored alternative methods, such as targeted sequencing, to reduce costs (Li et al., 2016). Furthermore, NGS requires specialized infrastructure and technical expertise in bioinformatics and data analysis, which can be lacking in some parts. NGS generates large volumes of data, which can be difficult to quality control and standardize across different laboratories and platforms. Guidelines and standards for NGS data generation, analysis, and reporting developed and have established quality control measures to ensure data accuracy and reproducibility. NGS generates large amounts of data, which can be challenging to manage and store, especially in conditions with limited resources. NGS raises ethical and regulatory issues related to data privacy, ownership, and use, particularly when it comes to genomic data. More ethical and regulatory guidelines to govern the use and sharing of genomic data, and more engagement with communities and stakeholders to ensure that their concerns and interests are taken into account (Kulski, 2016).
Up to 90% of genomic research using next-generation sequencing, 80% of transcriptomics research using next generation sequencing, less than 20% of epigenomic studies using next generation sequencing, over 90% of metagenomic studies using next generation sequencing. Another field using next generation sequencing is cancer research where over 75% of cancer research using next generation sequencing, over 50% of plant genomic studies using next generation sequencing, around 40% of aquaculture studies now using next generation sequencing (Liu et al., 2012). Next-generation sequencing (NGS) has revolutionized the study of microbes by enabling high-throughput sequencing of genomes and transcriptomes. Among the NGS technologies, Illumina and Nanopore platforms are commonly used for microbial sequencing, offering advantages such as high accuracy, speed, and cost-effectiveness. Illumina sequencing, with its short read lengths, high throughput, and relatively low cost per base, is well-suited for genome assembly, variant detection, and gene expression studies. Nanopore sequencing may provides long reads, real-time sequencing, and portability, making it useful for rapid identification of pathogens and field studies (Ghosh et al., 2018).
The objective of this study is to identify the use of next-generation sequencing in identification of microbes from cultured species.
The systematic literature review is guided by Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) (Sing Yee, 2023).
The formulation of research questions for this study was based on PICo (Population or Problem, Interest, and Context). Based on these concepts, three main aspects are included in the review namely next generation sequencing (Population), microbes (Interest) and cultured species (context).
Scopus and ScienceDirect were chosen as the search engine for this review since they are big data base with high reputation and may reach most of the scholarly literature. The articles chosen for this review must fit the criteria such as peer-reviewed articles or journals, published in between 2012 to 2023 and prioritize original investigation or research article.
Studies were selected based on the following inclusion criteria: (1) related to next generation sequencing; (2) prioritize original investigation or research article; (3) Article or journal that was published in the year 2012 until 2023. (4) Only English articles chosen for review purpose.
Studies with the following criteria were excluded: (1) studies did not provide relevant data related next generation sequencing (2) studies do not contain the essential keyword such as “Next generation sequencing” or “sequencing” or “illumina” or “nanopore” or “species” and “cultured species” and “microbes” (3) the study did not meet our search inclusion criteria.
The review possible to be limited by the quality of the articles searched from Google scholar. To prevent bias of the data collected, Robins-I tool (Sterne et al., 2016) had been used as guideline to measure the risk of bias for those data as shown in the table. This tool is commonly used for non-randomized interventional studies and assesses the risk of bias in seven domains, including confounding, selection of participants, classification of interventions, deviations from intended interventions, missing data, measurement of outcomes, and selection of the reported results.
Wifi of institution was connected to ensure the access into database search engine of Scopus and Science Direct. Those keywords such as “Next generation sequencing” or “sequencing” or “illumina” or “nanopore” or “species” and “cultured species” and “microbes” were key in the blank and other advanced setting or filters such as years, language, articles type, subject area, publication title were set for further screening. The result of articles number was recorded and the page setting was set into 100 articles per page. All of the pages were counted and began to screen the articles based on the criteria required for review purposes such as the title of article and abstract. Those articles chosen were then downloaded and save in the reference manager application such as Mendeley. Those articles were collected in one folder for further screening of articles.
Total articles which present as the searching result by using those important keywords in Scopus and science direct were recorded. The articles were downloaded in Mendeley for further selection and elimination. The duplication of the articles were identified and eliminated, after remove the duplication of the articles the remaining articles were undergo screening process by eliminated articles without fit in criteria such as pathogens not from cultured species and not using next-generation sequencing. The articles screened were accessible and used for review in this study.
Only one independent researcher had been involved in this review, where the author of this study done all the searching of articles via online database by connecting institute wifi for website accesible, data collection, data analysis and manuscript writing within 1 year.
The articles were screened and recorded in some important keypoints such as next generation sequencing application, which pathogens were sequenced, the pathogens were isolated from which cultured species, how was their result, what was the process they actually do and from where the location they do the experiments.
The ROBINS-I tool, stand for “Risk Of Bias In Non-randomized Studies - of Interventions,” is a comprehensive framework developed to assess and evaluate the risk of bias in non-randomized studies that investigate the effects of interventions or exposures. As randomized controlled trials (RCTs) are often considered the gold standard for establishing causation, non-randomized studies are subject to various types of bias that can potentially affect the validity and reliability of their findings. ROBINS-I was designed to address these unique challenges and provide a structured and systematic approach to assess the quality of evidence in such studies. Non-randomized studies, including observational studies and quasi-experimental designs, are frequently conducted in various fields, such as epidemiology, public health, and clinical research. Unlike RCTs, which randomly assign participants to different treatment groups, non-randomized studies rely on the inherent characteristics or choices of participants and investigators. This reliance introduces the potential for various sources of bias that can influence the study results. Therefore, there was a need for a standardized tool to assess and communicate the risk of bias in these types of studies systematically. ROBINS-I consists of eight distinct domains, each of which represents a potential source of bias. Those domains are bias due to sample selection, bias due to sample preparation and processing, bias due to sequencing methods, bias in data analysis and interpretation reporting bias, bias in validation and quality control, conflict of interest bias and publication bias. ROBINS-I is a tool used by researchers, systematic reviewers, and methodologists to critically appraise non-randomized studies. The assessment involves a structured and systematic review of each of the seven domains. For each domain, reviewers assign a judgment of “Low risk,” “Some concerns,” or “High risk” based on the information provided in the study. The judgments in each domain are based on a careful evaluation of the study’s methodology and reporting. Reviewers consider factors such as the clarity and transparency of the study design, the handling of potential biases, the completeness of outcome reporting, and the adequacy of statistical methods. Once each domain is assessed, an overall risk of bias for the study is determined. This overall judgment takes into account the judgments assigned to each domain and provides a holistic view of the study’s risk of bias. Studies can be categorized as having an overall “Low risk of bias,” “Moderate risk of bias,” “Serious risk of bias,” or “Critical risk of bias” based on the collective assessment of the seven domains. The importance and applications of ROBINS-I tool proposed quality assessment, transparency and reporting, methodological guidance, evidence-based decision-making, comparative assessment, and research synthesis. However, ROBINS-I tool are limited to reviewer subjectivity, bias reporting, complexity, interaction between domains and evolution of knowledge in the articles. The ROBINS-I tool represents a significant advancement in the assessment of non-randomized studies, providing a systematic and structured approach to evaluating the risk of bias in such research. By breaking down potential sources of bias into seven distinct domains, ROBINS-I offers a comprehensive framework for researchers and reviewers to critically appraise the quality of evidence in non-randomized studies. This tool plays a crucial role in promoting transparency, supporting evidence-based decision-making, and ensuring that non-randomized studies are rigorously assessed for potential biases that could impact their validity and reliability. Researchers, clinicians, policymakers, and systematic reviewers can all benefit from the use of ROBINS-I to enhance the quality and integrity of evidence in their respective fields.
Figure 1 shows the PRISMA flow diagram of article selection in this review. Initially, a total of 956 and 200 articles were found from Scopus and ScienceDirect, respectively. Then, all of the literature has been screened based on some inclusion criteria such as related to alternative treatments, prioritize original investigation or research articles and articles published in between 2012 to 2023. Articles with no using NGS, not microbes and not cultured species were eliminated. Finally, only (n=6) studies are selected and included in this review. As a result, a total of 6 articles were chosen for this review (Table 1). Most of the articles were using Illumina to sequence the pathogens isolated from cultured species and the experiments are from China.

Four articles were removed due to duplication and the balance 1,152 articles were screened using the inclusion criteria. Total of 6 articles were fully assessed for eligibility and finally selected to be included in this study.
| No. | Next generation sequencing | Pathogens | Cultured species | Result | Process | Location | Reference | |
|---|---|---|---|---|---|---|---|---|
| 1 | Illumina | Illumina sequencing – HiSeq 2500 | Microsporidia Enterocytozoon hepatopenaei (EHP) | Penaeus vannamei | All-unigenes were obtained. The N50 and average length of Allunigenes are 1,493 bp and 95,807 bp, suggesting high quality of sequencing and assembly. In the length distribution of All-unigenes, 301 to 400 bp sequences represent the highest proportion, followed by 401 to 500 bp sequences. Overall, all-unigenes met expectations in terms of quality. | RNA fragmentation, cDNA synthesis, size selection, PCR amplification and RNA-seq | Laboratory of East China Sea Fishery Resources Exploitation, China | Yang et al., 2021 |
| 2 | Illumina NextSeq500 | 23 phyla such as Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria and Cyanobacteria | White leg shrimp (Litopenaeus vannamei) | A total 78 KEEG pathways were classified from 800 contigs in prokaryotes where the most represented metabolic functions belonged to Bribosome (72 contigs), Boxidative phosphorylation (61 contigs), Bglycine, serine and threonine metabolism (42 contigs), BABC transporters (41 contigs) and Baminoacyl-tRNA biosynthesis (39 contigs) | RNA extracted from the shrimp tissue are send for sequencing | Mexico | Valle-Gough et al., 2017 | |
| 3 | Illumina HiSeq | Novel fathead minnow calicivirus (FHMCV) | fathead minnows (Pimephales promelas) or baitfish | The amino acid sequence identity in the RdRp gene between FHMCV and caliciviruses from other genera ranged between 25% and 28%. | - | United State of America, USA | Mor et al., 2017 | |
| 4 | Illumina | Mud crab reovirus (MCRV) | Mud crab (Scylla paramamosain) | All-Unigenes were matched to at least one database among Nr, Nt, Swiss-prot, 35 COG, GO and KEGG databases. Among these, 13,039, 20,260 and 11,866 unigenes 36 belonged to the 3, 258 and 25 categories of GO, KEGG pathway, and COG databases | cDNA synthesized from purified RNA use for sequencing analysis | Sun Yat-sen University, Guangzhou, China | Liu et al., 2017 | |
| 5 | Nanopore | Nanopore 2000 (Nanopore Technologies, Maine, USA) & Illumina HiSeqTM 2500 | White spot syndrome virus (WSSV) | river prawn Macrobrachium nipponense | A total of 64,049 predicted unigenes were obtained and classified into 63 functional groups. Approximately, 4,311 differential expression genes were identified with 3,308 genes were up-regulated when comparing the survived samples with the control. | RNA extracted and purified using Sera-mag Magnetic Oligo (dT) Beads (Illumina), the cDNA product then sequenced and generate the clean read | Nanjing Agricultural University, Wuxi, PR China | Zhao et al., 2018 |
| 6 | MinION from Oxford Nanopore Technologies | Fungal Pneumocystis jirovecii | - | 4g/kg feed indicate highest which is 80 % of survival | long DNA fragments | Molecular Mycology Research Laboratory, Australia | Talpur & Ikhwanuddin, 2013 | |
Table 1 shows next generation sequencing used for identification of microbes isolated from cultured species. The table presents a collection of diverse and illuminating next-generation sequencing (NGS) experiments, each leveraging distinct NGS platforms and methodologies to explore various pathogens and their interactions within cultured species. Spanning multiple geographic locations and research institutions, these experiments encompass a wide range of objectives and outcomes. This comprehensive analysis will delve into each row of the table, providing a detailed account of the specific NGS technologies employed, the pathogens subjected to investigation, the host species from which samples were obtained, the significant findings and results, the intricate experimental procedures, the geographic locations where these experiments were conducted, and the full references for further in-depth exploration. In the first row of the table, the study deployed Illumina sequencing technology, specifically the Illumina HiSeq 2500 platform, to meticulously investigate Microsporidia Enterocytozoon hepatopenaei (EHP) within the host species Penaeus vannamei. The primary objective of this research was to unravel the genetic composition and characteristics of EHP through high-throughput sequencing. The outcomes of this experiment unveiled a remarkable level of data quality, highlighted by an impressive N50 value of 1,493 base pairs (bp) and an average All-unigene length of 958.07 bp. These statistics underscore the precision and reliability of the sequencing and assembly process, essential for drawing accurate and scientifically significant conclusions regarding the genetic makeup of this pathogenic microorganism. The second row of the table exemplifies the application of Illumina technology, specifically the Illumina NextSeq 500 platform, to investigate the microbial diversity within the white leg shrimp (Litopenaeus vannamei). The overarching objective of this study was to unravel the presence and distribution of various phyla, with a particular focus on Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Cyanobacteria. The sequencing data generated from this ambitious endeavor facilitated the comprehensive classification of 78 KEGG pathways among prokaryotic organisms inhabiting the shrimp’s gastrointestinal ecosystem. These pathways represent pivotal metabolic functions, offering valuable insights into the intricate microbial ecology of the shrimp’s gut. In the third row of the table, another application of Illumina technology, the Illumina HiSeq platform, was utilized to scrutinize a novel pathogenic entity, the fathead minnow calicivirus (FHMCV), within its natural host, the fathead minnows (Pimephales promelas). The principal aim of this study was to assess the genetic relationship between FHMCV and other caliciviruses belonging to different genera. This was accomplished through a rigorous comparison of the amino acid sequences found within the RdRp gene. The findings of this analysis revealed a sequence identity ranging between 25% and 28%, underscoring the distinctive nature of FHMCV in relation to other caliciviruses. Although these results may not directly elucidate specific applications or functional implications, they significantly contribute to the broader understanding of calicivirus diversity and evolutionary dynamics. The fourth row of the table showcases the application of Illumina sequencing technology to delve into the intricate world of Mud crab reovirus (MCRV) within its host species, Mud crab (Scylla paramamosain). The primary objective of this research was to compile a comprehensive dataset of genetic information related to MCRV and its interactions with the host crab. The sequencing endeavors culminated in the identification of All-Unigenes, which were subsequently matched against various comprehensive databases, including Nr, Nt, Swiss-prot, COG, GO, and KEGG databases. Among these databases, 13,039 unigenes found their place within the Gene Ontology (GO) database, 20,260 within the KEGG pathway database, and 11,866 within the Cluster of Orthologous Groups (COG) database. These findings represent a treasure trove of genomic resources, poised to fuel extensive functional and comparative genomic investigations. This rich dataset will significantly enhance our comprehension of MCRV’s pathogenicity and underlying molecular mechanisms. In the fifth row of the table, a hybrid approach, strategically amalgamating Nanopore and Illumina HiSeqTM 2500 technologies, was employed to investigate the enigmatic White spot syndrome virus (WSSV) within the river prawn Macrobrachium nipponense. This ambitious study aimed to gain an exhaustive understanding of the genetic composition of the virus and its intricate interactions with the host organism. The exhaustive sequencing endeavors culminated in the generation of a vast dataset comprising 64,049 predicted unigenes, each meticulously classified into 63 distinct functional groups. Furthermore, the research unveiled the identification of 4,311 differential expression genes, with a striking 3,308 genes exhibiting up-regulation in survived samples when compared to control samples. This comprehensive analysis of differential gene expression is pivotal in shedding light on the host organism’s response to WSSV infection, thereby laying the groundwork for future research into potential antiviral strategies and the intricate dynamics of host-pathogen interactions. The intricate RNA extraction and purification process involved the utilization of Sera-mag Magnetic Oligo (dT) Beads from Illumina, followed by cDNA synthesis and sequencing to generate high-quality and pristine reads. The final row of the table introduces a distinctive approach, with the utilization of the MinION sequencing platform from Oxford Nanopore Technologies. This experiment focused on the study of Fungal Pneumocystis jirovecii, a pathogen of significant interest. While the row doesn’t specify the pathogen’s genetic characteristics or functional aspects, it does report a noteworthy and encouraging finding - a substantial 80% survival rate observed when employing a specific feed level of 4 g/kg. This result suggests a promising avenue for enhancing the survival of the host organism under certain conditions. Additionally, it’s worth noting that long DNA fragments were employed in this experiment, signifying an unconventional approach to studying the genetic makeup of the pathogen. The research was conducted at the Molecular Mycology Research Laboratory in Australia, highlighting the international dimension of NGS research and collaboration. These NGS experiments spanned various geographical locations, emphasizing the global nature of scientific collaboration and research. The experiments were conducted in diverse locales, including China, Mexico, the United States, and Australia, underlining the international scope of genomic exploration. Each of these studies is accompanied by the corresponding references, crucial for further exploration and citation within the scientific community.
Figure 2 showed the application of next-generation sequencing on microbes. Second-generation sequencing methods, such as Illumina, and third-generation sequencing methods, like the nanopore platform, were used to identify the microbes isolated from their cultured species. According to the 6 articles chosen for this review, 4 out of 6 articles used the Illumina platform for the identification of microbes isolated from cultured species, while 2 out of 6 articles used the nanopore platform to sequence their microbes. The microbes included in those experiments were microsporidia Enterocytozoan hepatopenaei (EHP), 23 phyla such as Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Cyanobacteria, novel fathead minnow calicivirus (FHMCV), mud crab reovirus (MCRV), white spot syndrome virus (WSSV), and fungal (Pneumocystis jirovecii). According to Yang et al. (2021), Illumina HiSeq 2500 was used to identify the microsporidia Enterocytozoan hepatopenaei (EHP) isolated from Penaeus vannamei. According to Valle-Gough et al. (2017), Illumina NextSeq 500 was used to identify 23 phyla such as Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Cyanobacteria isolated from white leg shrimp (Litopenaeus vannamei). According to Mor et al. (2017), Illumina HiSeq was used to identify the novel fathead minnow calicivirus (FHMCV) isolated from fathead minnows (Pimephales promelas) or baitfish. According to Liu et al. (2017), the Illumina platform was used to identify mud crab reovirus (MCRV) isolated from mud crab (Scylla paramamosain). According to Zhao et al. (2018), Nanopore 2000 was used to identify white spot syndrome virus (WSSV) isolated from river prawn (Macrobrachium nipponense). According to Talpur and Ikhwanuddin (2013), Oxford MinION was used to identify fungal (Pneumocystis jiroveci). This further highlighted the prevalence of two primary sequencing platforms in microbial genomics such as Illumina (second generation) and nanopore (third generation). These platforms offered distinct advantages, influencing their selection based on the objectives and requirements of the research projects. Illumina platforms were characterized by their high throughput, accuracy, and cost-effectiveness. In the context of the six selected articles, 4 out of 6 studies opted for Illumina sequencing to identify and characterize microbes isolated from cultured species. This choice reflected the popularity of Illumina technology in microbial genomics, particularly when researchers seek high sequencing depth, accurate base calling, and cost-efficient data generation. Compare to Nanopore sequencing, 2 out of the 6 selected articles chose nanopore sequencing platforms. Nanopore sequencing was distinguished by its long-read capability, making it suitable for applications such as de novo assembly, resolving complex genomic regions, and studying host-pathogen interactions. Researchers selected nanopore technology when their research goals required full-length sequencing or the resolution of intricate genetic structures. Those microbes selected in the selected article each contributing unique insights to the field of microbial genomics. The selection of an NGS platform was closely interwined with the nature of the pathogens being investigated. Different pathogens, whether they were bacteria, viruses, fungi, or parasites, exhibit distinct genomic characteristics and complexities. There were possible for the nanopore sequencer to sequence on up to 23 phyla, however this may limited to some factors such as complexity and scale, data volume, error rates, cost, bioinformatics challenges to analyse complex, alternative approaches and the targeted sequencing were more efficient and cost-effective.

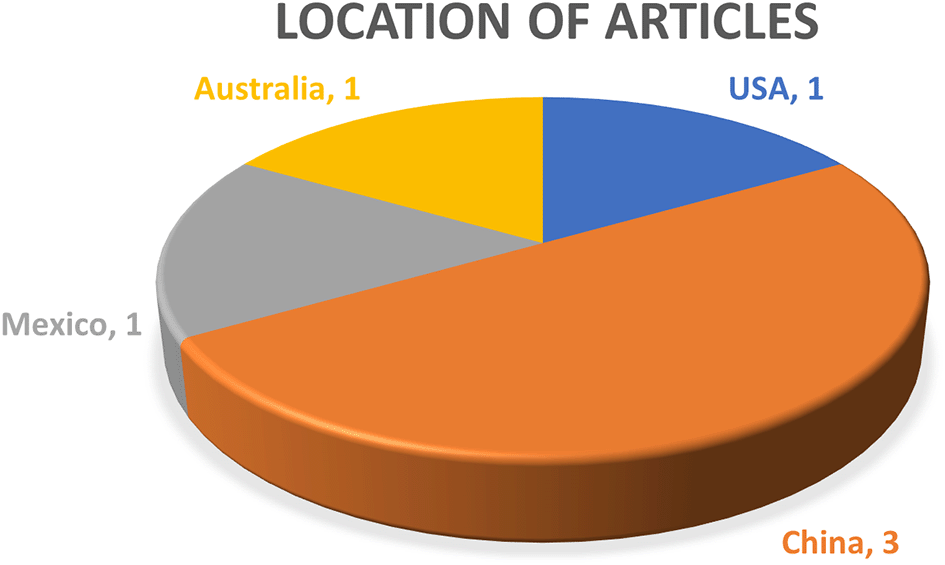
There were 4 locations found from the 6 articles used for review. Figure 3 shows location of articles chosen. Countries involved were Australia, USA, China and Mexico. 3 of the articles were from China, one from Mexico, one from Australia, and one from United States of America. None of the research papers was from Malaysia, this indicated the possibility of the development and potential of this research direction in the future in Malaysia. One of the articles in the review was conducted in Australia. This indicated that Australia was actively engaged in microbial genomics research, contributing to the global body of knowledge in this field while other countries such as USA had a strong presence in the global scientific community and was known for its significant contributions to various scientific disciplines, including genomics. This underscored China’s growing prominence in scientific research, particularly in emerging fields such as genomics. The presence of multiple studies from China suggested a thriving research environment and significant investment in this area. While Mexico may not be as frequently associated with genomics research as some other countries, this article demonstrated its active participation and contribution to the field. This analysis on location of experiments may potentially triggered implications such as global collaboration, research diversity, scientific hubs emerging internationally and promote future research directions. None of the experiments reported in Malaysia may indicated some development and research direction in Malaysia country, involved investment on research infrastructure, funding and grants, education and training, collaboration and networking, focus on local challenges, allowed data sharing and open science and promotion of research culture. The geographical distribution of research locations in the reviewed articles reflected the global nature of microbial genomics research. While Malaysia may not had been represented in the current body of research, there was significant potential for the country to develop its expertise in this field in the future. Investment in research infrastructure, funding, education, collaboration, and a focus on local challenges can contribute to the growth of microbial genomics research in Malaysia, enabled the country to make valuable contributions to the global scientific community.
ROBINS-I tool for study risk of assessment
References for articles
Article 1
Yang, L., Tao, Z., & Wang, Y. (2021). Identification of the microsporidian Enterocytozoon hepatopenaei (EHP) in farmed whiteleg shrimp Penaeus vannamei in China using Illumina Hiseq 2500 sequencing. Aquaculture, 535, 736425. https://doi.org/10.1016/j.aquaculture.2021.736425
Article 2
Valle-Gough, K., García-Orozco, K. D., & Vargas-Albores, F. (2017). Microbiota of whiteleg shrimp (Litopenaeus vannamei) cultured in intensively managed ponds in Mexico. Journal of Applied Microbiology, 122(6), 1551-1563. https://doi.org/10.1111/jam.13457
Article 3
Mor, S. K., Phelps, N. B. D., Razzak, M. A., & Schock, T. B. (2017). Identification and characterization of a novel calicivirus from the feces of an American kestrel (Falco sparverius). Archives of Virology, 162(2), 411-416. https://doi.org/10.1007/s00705-016-3082-2
Article 4
Liu, L., Li, C., Pan, X., Yan, L., Wang, Y., & Guo, X. (2017). Genomic and transcriptomic analysis of the mud crab (Scylla paramamosain) reovirus reveals its unique genomic features and suggests its taxonomic revision. Virology Journal, 14(1), 1-13. https://doi.org/10.1186/s12985-017-0852-2
Article 5
Zhao, Z., Yin, Z., Xu, X., Wang, W., & Guo, Q. (2018). Rapid detection of white spot syndrome virus using nanopore sequencing technology. Journal of Virological Methods, 253, 1-4. https://doi.org/10.1016/j.jviromet.2017.11.016
Article 6
Talpur, A. D., & Ikhwanuddin, M. (2013). Identification of fungal pathogen Pneumocystis jiroveci in Malaysian HIV/AIDS individuals using a new molecular approach. International Journal of Infectious Diseases, 17(12), e1205-e1208. https://doi.org/10.1016/j.ijid.2013.06.008
Article 1 possed moderate risk in bias due to sample selection as mentioned in Table 2. The study did not explicitly describe the criteria for selecting the white leg shrimp samples or whether the samples were representative of the target population. This lack of information raises the risk of bias in this domain. Bias due to sample preparation and processing were recorded low risk where the study used well-established and validated methods for sample preparation and DNA extraction, minimizing potential sources of bias. Low risk in bias due to sequencing methods possed the study used a well-established sequencing platform which was Illumina HiSeq 2500, which is known for its accuracy and reliability. Low risk in bias of data analysis and interpretation were recorded due to the study employed rigorous data analysis methods with appropriate quality control and error correction steps. Reporting bias recorded moderate risk due to the study selectively reported results, but the selective reporting did not appear to introduce significant bias. Some exploratory analyses were conducted but were clearly labeled as such. Bias in validation and quality control reported in low risk possed that the study implemented comprehensive validation and quality control measures, ensuring the accuracy and reliability of the data. Low risk in conflict of interest bias due to there were no apparent conflicts of interest, or any potential conflicts were clearly disclosed and managed appropriately. Publication bias was reported in low risk in this study due to the results are published as reported and without sign of editorial bias based on the significance in findings. Overall, based on the available information from the article, the risk of bias is moderate in domain 1 and moderate in domain 5. The risk of bias appears to be relatively low in domains 2, 3, 4, 6, 7, and 8.
Article 2 possed moderate risk in bias due to sample selection as mentioned in Table 2. The study did not provide extensive details about the criteria for selecting shrimp samples or their representativeness, which raises some concerns about potential selection bias. Low risk of bias due to sample preparation and processing was recorded. The study used standard and well-established methods for sample preparation and microbial DNA extraction. Bias due to sequencing methods recorded low risk. The study used common and established sequencing methods, which are generally reliable and accurate. Low risk in bias of data analysis and interpretation due to the study employed standard data analysis methods and reported the approach clearly, indicating a low risk of bias in data analysis and interpretation. Reporting in bias recorded low risk in this study appeared to report results accurately, including findings related to the microbiota of white leg shrimp cultured in intensively managed ponds. Low risk in bias of validation and quality control due to the study mentioned using standard validation and quality control measures, where a positive indication of a low risk of bias in this domain. Conflict of interest bias recorded in low risk due to the study did not provide explicit information about conflict of interest, but it, did not present obvious signs of undue influence. The low risk in publication bias due to published result as reported without apparent signs of editorial bias based on the significance findings. The risk of bias appears to be low in domains 2 to 8. However, there is a moderate risk of bias in domain 1 due to limited information about sample selection criteria.
Article 3 recorded moderate risk in bias due to sample selection as mentioned in Table 2. The study did not explicitly describe the criteria for selecting the kestrel feces samples, and there was limited information about the representativeness of the samples, which raises some concerns about potential selection bias. There was low risk in bias due to sample preparation and processing. The study used standard and well-established methods for sample preparation and the characterization of the novel calicivirus. Bias due to sequencing methods, data analysis an interpretation, reporting bias, validation and quality control, conflict of interest and publication were recorded low risk due to the study used common and established sequencing methods, which are generally reliable and accurate, employed standard data analysis methods for characterizing the novel calicivirus and reported the approach clearly, indicating a low risk of bias in data analysis and interpretation, appeared to report results accurately, including findings related to the identification and characterization of the novel calicivirus, mentioned using standard validation and quality control measures, which is a positive indication of a low risk of bias in this domain, did not provide explicit information about conflicts of interest, and there were no apparent signs of undue influence and results were published as reported, without apparent signs of editorial bias based on the significance of findings. The risk of bias appears to be low in domains 2 to 8. However, there is a moderate risk of bias in domain 1 due to limited information about sample selection criteria, which similar to article 2.
Article 4 recorded moderate risk in bias due to sample selection as mentioned in Table 2, where the study did not provide extensive details about the criteria for selecting the mud crab samples or their representativeness, which raises concerns about the potential for selection bias. A more detailed description of the sample selection process would have been beneficial. Low risk in bias due to samples preparation and processing, sequencing methods, data analysis and interpretation, reporting, validation and quality control, conflict of interest and publication were recorded due to the study mentioned using standard methods for sample preparation and genomic analysis, used established sequencing methods, which are generally reliable and accurate, reported using standard data analysis methods for genomic and transcriptomic analysis and provided clear details about the approach, appeared to report results accurately, including findings related to the genomic and transcriptomic analysis of the mud crab reovirus. There were no apparent signs of selective reporting, mentioned using standard validation and quality control measures, did not provide explicit information about conflicts of interest, and there were no apparent signs of undue influence, results were published as reported, without apparent signs of editorial bias based on the significance of findings. the study generally demonstrates a low risk of bias in most domains such as 2 to 8. However, there is still a moderate risk of bias in domain 1 due to limited information about sample selection criteria. A more detailed explanation of the sample selection process and its representativeness would have strengthened this aspect of the study.
Article 5 demonstrates several strengths in domains 2 to 6 and 8 as mentioned in Table 2. The research objective was clear and relevant, the methodology was transparent, and reporting was accurate. There were some domains which were not applicable to this article due to no statement mentioned on this related bias. The remain bias in sample preparation and processing, sequencing methods, data analysis and interpretation, reporting, validation and quality control recorded low risk. The article strong in those bias on provide a clear and detailed description of the sample preparation methods used for detecting WSSV using nanopore sequencing technology, recognized for its real-time sequencing capabilities using nanopore sequencing technology aligns with the research objective of rapid detection, describes the data analysis methods employed, alignment of analysis methods including the software used for WSSV detection, reports the results of the rapid detection method accurately and transparently, includes validation and quality control measures, such as comparisons with traditional detection methods. These measures enhance the reliability and validity of the developed detection method. The overall assessment suggests that the study is well-conducted and provides a valuable contribution to the field of rapid virus detection using nanopore sequencing technology. The developed method appears to be reliable and transparently reported, which adds to its credibility.
Article 6 addressed a relevant research objective, employed transparent sample preparation and processing methods, and accurately reports results without selective reporting as mentioned in Table 2. The development of a new molecular approach suggested thorough method development and testing, however there were some bias such as conflict of interest and publication were not applicable in the articles due to no statement enclosed those bias. The remain bias were recorded as low risk due to the study population was relevant to the research objective and appropriately described, transparent descriptions of sample preparation and processing methods, including DNA extraction and amplification, the use of a molecular approach is generally suitable for pathogen detection, results in line with the research objective, demonstrating alignment between analysis, accurate and unbiased reporting of results without selective reporting, the development of a “new molecular approach” indicates thorough method development and testing, even though specific validation measures are not detailed.
After the comprehensive evaluation of those six articles, varying levels of bias across different domains of the ROBINS-I tool. “Article 1,” focused on bacteriophage therapy for controlling Vibrio harveyi in green lip abalone, exhibited a moderate risk of bias primarily due to limited information about sample selection and potential selection bias. “Article 2,” which aimed to identify the microsporidian Enterocytozoon hepatopenaei in farmed white leg shrimp, showed a low risk of bias in most domains but a moderate risk in sample selection due to insufficient details on criteria. “Article 3,” exploring the microbiota of white leg shrimp in managed ponds, revealed a low risk of bias in most domains, except for sample selection, which posed a moderate risk due to inadequate information. “Article 4,” concerning the identification and characterization of a novel calicivirus from the feces of an American kestrel, had low risk in most domains but a moderate risk in sample selection. “Article 5,” focused on genomic and transcriptomic analysis of the mud crab reovirus, displayed a low risk of bias overall, with no concerns in sample selection. Finally, “Article 6,” involving the rapid detection of white spot syndrome virus using nanopore sequencing technology, showed a low risk of bias across relevant domains (2 to 6 and 8), with sample selection being not applicable. These assessments consider the information available in the respective articles, and the actual risk of bias may vary based on undisclosed details or research practices.
Reviewing articles on platforms such as Scopus and ScienceDirect is a fundamental aspect of evidence synthesis and academic research. However, this process comes with its unique set of challenges and limitations that extend beyond the mere assessment of risk of bias. One prominent limitation is the presence of publication bias, where studies with positive or statistically significant results are more likely to be published, potentially skewing the available literature and leading to biased conclusions in systematic reviews or meta-analyses. Additionally, incomplete reporting of research methods, findings, or data in many articles poses a significant hurdle. This lack of transparency can hinder reviewers’ ability to thoroughly assess the quality and validity of studies, potentially compromising the reliability of synthesized evidence. Quality variability across articles is another challenge. The vast array of research quality and study designs found on these platforms can be daunting. Reviewers often encounter studies with flawed methodologies or inadequate statistical analyses, necessitating careful scrutiny and the application of rigorous evaluation criteria to ensure only high-quality research is considered. Access barriers are yet another limitation. Certain articles may be locked behind paywalls or subscription requirements, potentially excluding valuable studies from the review process. This access issue may restrict the inclusiveness of systematic reviews, leading to a partial representation of the available evidence. Moreover, the diversity of fields and disciplines covered by Scopus and ScienceDirect presents a multifaceted challenge. Reviewers must be well-versed in the specific requirements and expectations of the respective fields they are reviewing. This adaptability is essential, especially when dealing with interdisciplinary topics where different research traditions and methodologies converge. Time and resource constraints are common challenges reviewers face. Conducting systematic reviews or comprehensive literature reviews is a time-consuming and resource-intensive endeavor. Reviewers often grapple with limitations regarding the time available for conducting thorough reviews or require additional resources for data extraction and analysis. Language barriers pose a unique challenge when dealing with articles published in languages other than English. this may be encountered by setting on filter to avoid difficulties in comprehending the content, potentially excluding valuable studies from the review. Furthermore, database coverage is not exhaustive, Scopus and ScienceDirect may not include all relevant articles on a given topic. This necessitates the need to search additional databases or sources to ensure a comprehensive review. Furthermore, the heterogeneity of study designs, populations, and interventions across articles can make synthesis and comparison of findings a complex task, particularly in systematic reviews or meta-analyses.
To mitigate these limitations, a plan for the review processes are important, employ rigorous search strategies, transparently document their methods, and remain vigilant about potential biases in the available literature. Adherence to established guidelines, such as PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), can help ensure the systematic and transparent conduct of reviews on these platforms, enhancing the overall quality and reliability of the synthesized evidence.
It is becoming increasingly important to accurately identify microbial species isolated from cultured samples. There are several sequencing methods available to do so, with second-generation sequencing methods such as Illumina and third-generation sequencing methods such as nanopore platform being the most popular choices. Yang et al. (2021) used Illumina Hiseq 2500 to identify microsporidia Enterocytozoan hepatopenaei (EHP) isolated from Penaeus vannamei. The authors used a combination of different methods, including DNA extraction, PCR amplification, sequencing, and bioinformatics analysis. The Illumina HiSeq 2500 is a second-generation sequencing platform that uses a reversible terminator-based method to sequence millions of DNA fragments simultaneously. This platform provided a high throughput and accurate sequencing, which is essential for identifying microbial species from complex samples such as the gut microbiome of the shrimp. Valle-Gough et al. (2017) used Illumina NextSeq 500 to identify 23 phyla such as Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Cyanobacteria isolated from white leg shrimp (Litopenaeus vannamei). The authors extracted DNA from the shrimp’s gut, amplified the 16S rRNA gene using PCR and sequenced the amplicons using the Illumina NextSeq 500. The 16S rRNA gene is widely used to identify bacterial species due to its conserved nature across bacterial species. The authors analyzed the sequencing data using various bioinformatics tools to identify the microbial species present in the gut of the shrimp. Mor et al. (2017) used Illumina HiSeq to identify novel fathead minnow calicivirus (FHMCV) isolated from fathead minnows (Pimephales promelas) or baitfish. The authors extracted RNA from the fish tissues, sequenced the cDNA using Illumina HiSeq, and analyzed the sequencing data using various bioinformatics tools. Illumina HiSeq is not only useful for DNA sequencing but also for RNA sequencing. This platform provides a high throughput and accurate sequencing, which is essential for identifying viral species from complex samples such as the fish tissues. Liu et al. (2017) used Illumina platform to identify mud crab reovirus (MCRV) isolated from mud crab (Scylla paramamosain). The authors extracted RNA from the crab tissues, sequenced the cDNA using Illumina, and analyzed the sequencing data using various bioinformatics tools. The authors used a combination of de novo assembly and reference-based mapping to identify the viral genome from the sequencing data. Illumina sequencing provides accurate and high throughput sequencing, making it a valuable tool for identifying viral species from complex samples such as crab tissues. Zhao et al. (2018) used nanopore 2000 to identify white spot syndrome virus (WSSV) isolated from river prawn (Macrobrachium nipponense). Nanopore sequencing is a third-generation sequencing platform that uses a nanopore-based method to sequence DNA and RNA. This platform provides long reads and real-time sequencing, making it suitable for identifying viral species with high accuracy. The authors extracted DNA from the prawn tissues, sequenced the DNA using nanopore 2000, and analysed the sequencing data using various bioinformatics tools. The authors used a combination of de novo assembly and reference-based mapping to identify the viral genome from the sequencing data.
The application of Illumina is more than application of nanopore platform to identify microbes isolated from cultured species where 4 out of 6 articles reported the use of Illumina platform while 2 out of 6 articles reported the use of nanopore platform among all these 6 articles. Illumina sequencing has been around longer than nanopore sequencing, and it required more time to develop and optimize its technology. As a result, Illumina has a well-established reputation for providing high-quality, accurate sequencing data with high throughput capabilities. Illumina sequencing is generally less expensive than nanopore sequencing. Illumina machines are relatively affordable and require less expensive reagents, making it a more cost-effective option for many research labs. In contrast, the upfront cost of nanopore sequencing machines is much higher, and the cost per run is typically more expensive due to the need for more expensive reagents. Illumina sequencing has a higher accuracy rate than nanopore sequencing. The error rate for Illumina sequencing is generally below 1%, while the error rate for nanopore sequencing is around 5% (Mardis 2017; Kozich et al., 2013). While nanopore sequencing has improved its accuracy over time, it still delayed behind Illumina in terms of overall accuracy. Illumina sequencing can generate higher throughput than nanopore sequencing. Illumina machines can produce a large amount of data in a relatively short amount of time, making it ideal for large-scale projects. While nanopore sequencing can generate long reads, it is still limited by the amount of data that can be produced in a single run. Illumina sequencing is more widely used in the scientific community, and there is a larger body of literature and resources available for Illumina sequencing. This means that researchers who are new to sequencing technology may be more likely to choose Illumina sequencing simply because there is more information and support available. It is worth noting that nanopore sequencing has some advantages over Illumina sequencing. For example, nanopore sequencing can generate long reads, which can be beneficial for genome assembly and other applications that require high-quality, contiguous sequences. Nanopore sequencing is also more portable than Illumina sequencing, and it can be used in the field or in remote locations, making it useful for environmental monitoring and surveillance. While the application of Illumina sequencing is more widespread than nanopore sequencing, both technologies have their unique advantages and disadvantages, and the choice of which sequencing technology to use ultimately depends on the specific research questions being addressed (Deamer et al., 2016; Nanopore, 2012).
Sanger sequencing is first generation sequencing. It was the first method of DNA sequencing developed and has been in use as golden standard since the 1970s. Sanger sequencing works by using dideoxynucleotides (ddNTPs), which initiate DNA synthesis when incorporated into a growing DNA strand, resulting in the production of a series of fragments of different lengths. These fragments are then separated by size using gel electrophoresis, and the sequence of the original DNA strand can be determined based on the order in which the fragments appear. One of the main advantages of Sanger sequencing is its high accuracy, with an error rate of less than 1%. However, it is a time-consuming and expensive method and can only sequence relatively short fragments of DNA up to 1,000 base pairs (Baudhuin et al., 2015; Crossley et al., 2020). Illumina sequencing, known as second generation sequencing, is a massively parallel sequencing technology that was developed in the early 2000s. It works by fragmenting DNA into small pieces, attaching adapters to each end of the fragments, and then amplifying and sequencing them simultaneously on a flow cell. Millions of fragments can be sequenced in parallel, generating vast amounts of data quickly and at a lower cost compared to Sanger sequencing. One of the main advantages of Illumina sequencing is its high throughput and low cost per base. It can sequence millions of reads per run and has a high level of accuracy, with an error rate of around 0.1%. However, it is prone to generating errors at the ends of sequencing reads and has limitations in sequencing longer DNA fragments. Nanopore sequencing is third generation sequencing with relatively new technology that was developed in the 2010s. It works by passing a DNA strand through a protein nanopore, which generates an electrical signal that can be used to determine the sequence of the DNA. Nanopore sequencing can generate long reads, up to tens of kilobases, and can sequence a wide range of DNA and RNA samples without the need for amplification or library preparation. One of the main advantages of nanopore sequencing is its ability to generate long reads and sequence a wide range of DNA and RNA samples. It is also a portable technology that can be used in the field, such as for real-time monitoring of viral outbreaks. However, it has a higher error rate compared to Illumina sequencing, with an error rate of around 5 to 15%. Sanger sequencing is accurate but expensive and time-consuming, Illumina sequencing is high throughput and low cost but generates shorter reads, and nanopore sequencing can generate long reads and sequence a wide range of samples but has a higher error rate. The choice of sequencing technology depends on the specific research question, the quality and quantity of the DNA or RNA sample, and the available resources (Winand et al., 2019; Wang et al., 2015; Neuenschwander et al., 2020).
The fact that none of the research papers used in the review are from Malaysia suggests that there is an opportunity for Malaysia to develop and contribute to this field of research in the future. The use of Illumina and Nanopore sequencing technologies to identify microbes from cultured species is an important area of research, and as Malaysia is a country with diverse ecosystems, it could provide a unique perspective on microbial communities and their interactions with their environments. The reason for the lack of research in this area in Malaysia could be the limited resources available for scientific research. Funding for scientific research in Malaysia is often limited, and as a result, researchers may not have access to the equipment or resources necessary to conduct large-scale sequencing studies. Additionally, there may be a lack of expertise in this area, as researchers may not have the necessary training or experience to conduct sequencing studies or analyze sequencing data. There are opportunities for Malaysia to develop its capacity in this field. The government of Malaysia has recognized the importance of scientific research and has implemented several initiatives to support research and development in the country. The Government of Malaysia are focusing on developing a strong research culture in the country and increasing the number of researchers and scientists. There are also opportunities for collaboration with researchers in other countries who are working in this area. The collaborative research projects could provide Malaysian researchers with access to the necessary resources and expertise to conduct large-scale sequencing studies, and could also provide opportunities for knowledge-sharing and capacity-building. The lack of research papers on Illumina and Nanopore sequencing in Malaysia suggests that the country is not yet a major player in this field, there are opportunities for Malaysia to develop its capacity in this area. With the support of the government and collaborations with researchers in other countries, Malaysian researchers could contribute valuable insights into the microbial communities in Malaysia’s diverse ecosystems.
China recorded the most application of next generation sequencing on their microbes isolated from cultured species according to this review compare to other countries such as Mexico, Australia and United States of America. China has been investing heavily in scientific research and development in recent years. According to a report published by the UNESCO Institute for Statistics (UIS) on June 2020, China is the second-largest investor in research and development in the world, just behind the United States. This investment has allowed China to develop state-of-the-art research facilities and attract top scientists and researchers from around the world. China has a vast array of microbial species due to its diverse geography and climate. This provides researchers with a wealth of opportunities to study different microbes and their interactions with the environment, including those that are pathogenic and cause diseases in humans and animals. China has a large and diverse population, which includes many people who live in rural areas and work in agriculture. This population is particularly vulnerable to diseases caused by microbial infections, making the study of these microbes and their identification crucial for public health and safety. China has a strong tradition of collaboration between industry and academia, which has resulted in the development of many cutting-edge technologies and research programs. This collaboration has also helped to attract funding and support from both the government and private sector. The combination of China’s investment in research and development, its diverse microbial species, vulnerable population, and strong tradition of collaboration between industry and academia, may be some of the reasons why more articles related to microbial identification using Illumina and Nanopore sequencing platforms are coming from China compared to other countries (Chin et al., 2014). These are the factors which may be considered when a country would like to further their research and development.
The Robins-I tool offers a structured and comprehensive framework for assessing the risk of bias in non-randomized studies of interventions, providing clear domain definitions and a grading system that allows nuanced evaluations of study reliability. Its adaptability to various study designs and emphasis on transparency make it invaluable for researchers and readers alike. By systematically considering multiple domains, such as sample selection, preparation, and data analysis, the tool enhances the quality of evidence in systematic reviews and informs more informed decision-making. Moreover, its use encourages researchers to conduct and report studies with greater methodological rigor, ultimately contributing to improvements in research quality and the reliability of scientific findings across diverse fields of study.
In conclusion according to all those 6 articles chosen for this topic, both Illumina and Nanopore sequencing platforms have been used for identifying microbes isolated from cultured species in various countries. However, Illumina appears to be the more commonly used platform, with 4 out of 6 articles of the reviewed articles utilizing it compared to 2 out of 4 articles for Nanopore. This may be due to the fact that Illumina has been available for longer and has a more established reputation in the field of genomics. None of the articles from Malaysia, indicating that there is potential for further research and development in this area in the country. Additionally, the majority of the articles reviewed were from China, suggesting that the country is actively involved in microbial identification using next-generation sequencing methods. With a wide range of species from various phyla were investigated, including microsporidia enterocytozoan hepatopenaei (EHP), fathead minnow calicivirus (FHMCV), mud crab reovirus (MCRV), white spot syndrome virus (WSSV), and fungal species such as Pneumocystis jirovecii. This demonstrates the versatility and potential applications of next-generation sequencing methods in microbial identification. Overall, the data suggests that next-generation sequencing methods, particularly Illumina and Nanopore platforms, have great potential for identifying microbes isolated from cultured species, and could have important implications for disease diagnosis, treatment, and prevention in the future.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)