Keywords
transcriptomics, differential gene expression, RNA-seq, heart development, cardiomyocyte differentiation, clustering, shiny application
transcriptomics, differential gene expression, RNA-seq, heart development, cardiomyocyte differentiation, clustering, shiny application
Differential gene expression analysis of developmental processes during embryogenesis can be enhanced by combining online resources that provide curated data from the scientific literature with independent investigations of previously published data. Several online databases exist, such as the Mouse Genome Informatics (MGI) online database, which aggregates in situ hybridization gene expression data (Genepaint), high-throughput gene expression datasets profiling a diverse set of tissues (Genotype-Tissue Expression (GTEx) Project), and the biocuration of gene expression data from diverse sources integrating anatomy and expression.1–3 However, the expression profiles of many genes remain ambiguous or poorly characterized during the spatiotemporal expression changes that occur during organogenesis. For example, according to the MGI Gene Expression Database (GXD), only 8,660 of the 16,038 genes identified by the resource have annotated gene expression data for the developing heart at any stage or expression level, using conventional molecular techniques.3,4 To fill these gaps in gene expression knowledge, the large quantity of high-throughput gene expression data aggregated within the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) provides an incredible opportunity for investigators to enhance their investigation of differential gene expression during developmental processes and thereby obtain preliminary data to develop new hypotheses for future investigations.
Data analyses involving two experimental groups are relatively straightforward to complete using GEO2R, a differential analysis software integrated into GEO.5 However, an in-depth study of the differentiation and development of a tissue or organ involves analysis of multiple experimental conditions comprising a developmental time series. Several bioinformatics tools and the requisite programming experience are required to complete the appropriate differential gene expression analysis and data visualization. We introduced developmental Gene Expression Analysis (devGEA), a new software solution that combines common bioinformatics tools necessary to investigate gene expression changes during cellular differentiation or other developmental processes, with an emphasis on classifying unique gene expression signatures. The workflow provides an investigator with the ability to evaluate and classify gene expression changes in a developmental or time-series dataset, such as differentiation from a pluripotent cell to a terminally differentiated cellular state. DevGEA extends the common steps of raw data processing by introducing several methods for analysis and classification based on a gene model or specific developmental gene expression. These methods are direct, reproducible, and conceptually simpler than many other clustering methods often used in this area. devGEA can be run in the browser from http://cs.indstate.edu/devgea or downloaded as an R source code from https://gitlab.com/jkinne/devgea and executed locally. The Gitlab repository also contains a standalone R Markdown file that performs the analysis in this study.
To demonstrate the use of this software tool, we used devGEA to perform differential gene expression analysis and developmental gene signature classification of cardiomyocyte differentiation from human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESC) and human induced pluripotent stem cell (hiPSC) lines. The two prior datasets utilized in our investigation profile the cardiomyocyte differentiation of several cell lines, providing an opportunity to classify cardiac developmental signature genes that are reproducible across multiple cell lines. Genes identified within these developmental signatures were validated in the laboratory for comparable expression changes in an independent hiPSC line.
hPSCs have revolutionized biology, allowing in vitro investigation of developmental, physiological, and pathological processes within a human tissue-based experimental system.6,7 The study of heart development and disease has undergone a paradigm shift in the last decade, evolving from the use of experimental organisms as models of mammalian heart development, such as the mouse, to the use of in vitro hPSC differentiation procedures to directly investigate human cardiac development.8–10 Furthermore, these breakthroughs have enabled scientists to study human cardiomyocyte differentiation and the physiology of hiPSCs in both healthy patients and those with congenital heart defects. Directed differentiation of hESCs or hiPSCs using either recombinant growth factors or small-molecule modulators of the WNT signaling pathway enables robust and efficient differentiation of hPSCs into cardiomyocytes, which recapitulates the differentiation of the cardiac lineage in vivo.11–14 Similar to embryonic heart development, hPSCs undergo mesoderm, cardiac progenitor, and cardiomyocyte differentiation, which are characterized by the stepwise expression of specific developmental mesodermal and cardiac regulatory genes.8,9,11 In this study, the devGEA analysis suite was used to classify gene expression profiles during human cardiomyocyte differentiation based on known cardiac developmental regulators. Together, these results aid our understanding of discrete gene expression profiles representing the major gene expression states of cardiomyocyte differentiation, and form a robust method to investigate novel cardiac genes.
in vitro directed cardiomyocyte differentiation using small molecule modulators of the WNT signaling pathway has greatly enhanced our ability to study human cardiomyocyte development.13,15,16 This procedure offers a robust and reproducible experimental assay for studying cardiomyocyte differentiation that recapitulates the in vivo cardiac developmental gene regulatory network. Compared to previous methods, such as embryoid body generation, which produces a heterogeneous mixture of developing tissues, directed cardiomyocyte differentiation enables the generation of cardiomyocytes at an efficiency varying from 80 to 98%.8,15,16 Since directed cardiomyocyte differentiation involves the introduction of defined culture media, developmental gene expression changes can be correlated with the introduction of new media formulations containing specific small molecules and supplements. These datasets consist of at least four developmental time points that correspond to specific culture media that induce a stepwise cardiomyocyte differentiation program. Standard bioinformatic analyses using pairwise statistical tests and fold-change cutoff criteria between groups can be unwieldy and difficult to interpret. Thus, differential gene expression analyses of such datasets have traditionally used statistical tools consisting of the ANOVA statistical test and pair-wise fold-change cutoff criteria, followed by data visualization using clustering tools such as k-means or hierarchical clustering. Given our prior knowledge of key regulatory genes within the cardiac network, a guided analytical approach can instead be employed to classify cardiac genes based on their unique developmental gene expression signatures. Figures 1 and 2 summarize the key timepoints of the cardiomyocyte differentiation experiment: the two prior datasets that we used for analysis and expression patterns of key genes from our study.
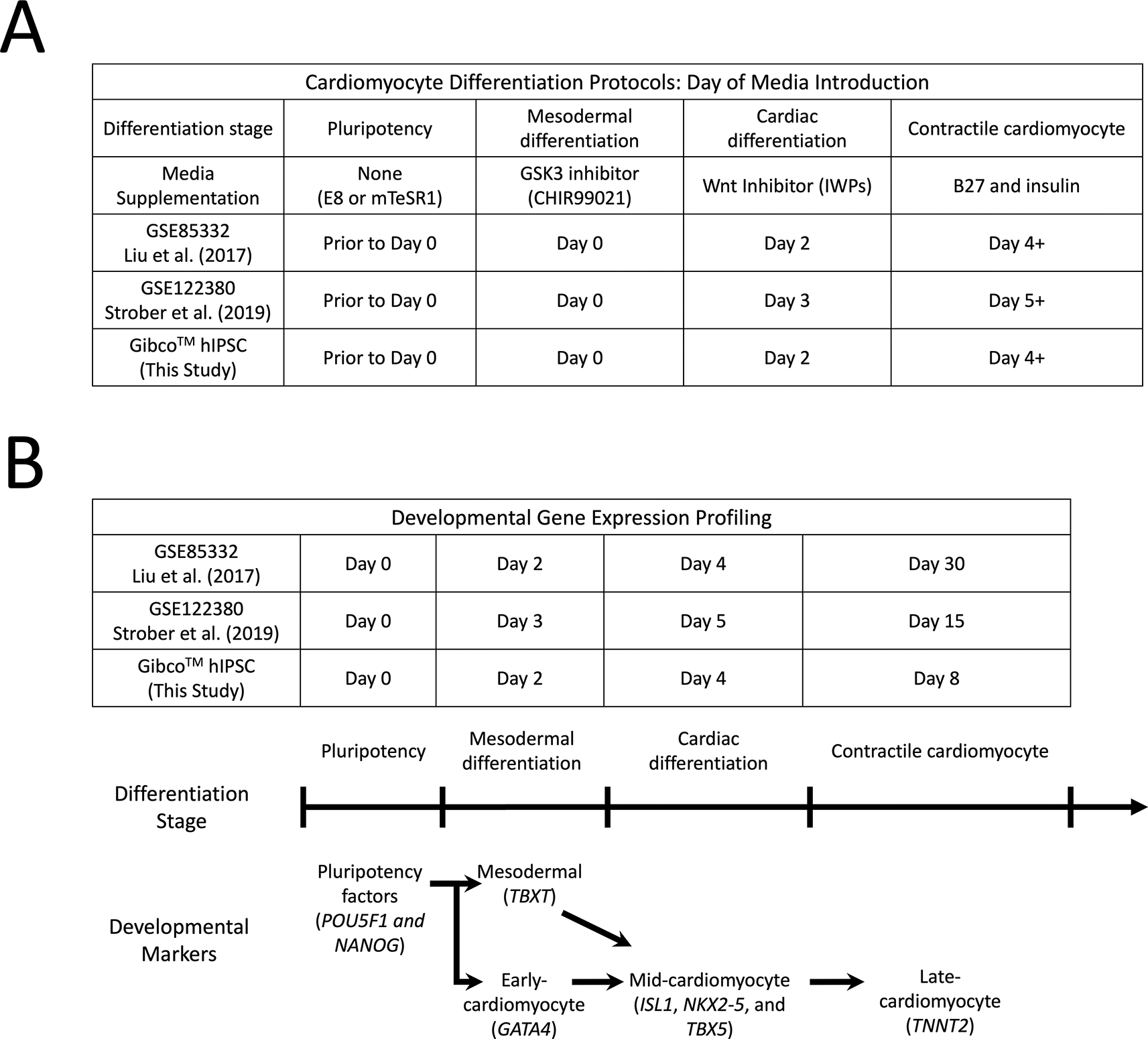
A) Methods of small molecule-directed cardiomyocyte differentiation of hiPSCs in prior studies and the present study. Wnt signaling pathway modulators are utilized in a stepwise manner to induce cardiomyocyte differentiation and to maintain contractile cardiomyocytes over the course of several days. Although the addition of specific small-molecule inhibitors and supplements varies slightly, the overall result efficiently generated a large population of autorhythmic contractile cardiomyocytes.
B) Gene expression profiling of three experimental datasets used in this study. The major developmental time points were identified along the cardiac differentiation stage timeline with comparable dates of gene expression profiling in each experiment identified in the table above. Key cardiomyocyte gene expression signatures as well as the expected cardiac developmental markers were identified.
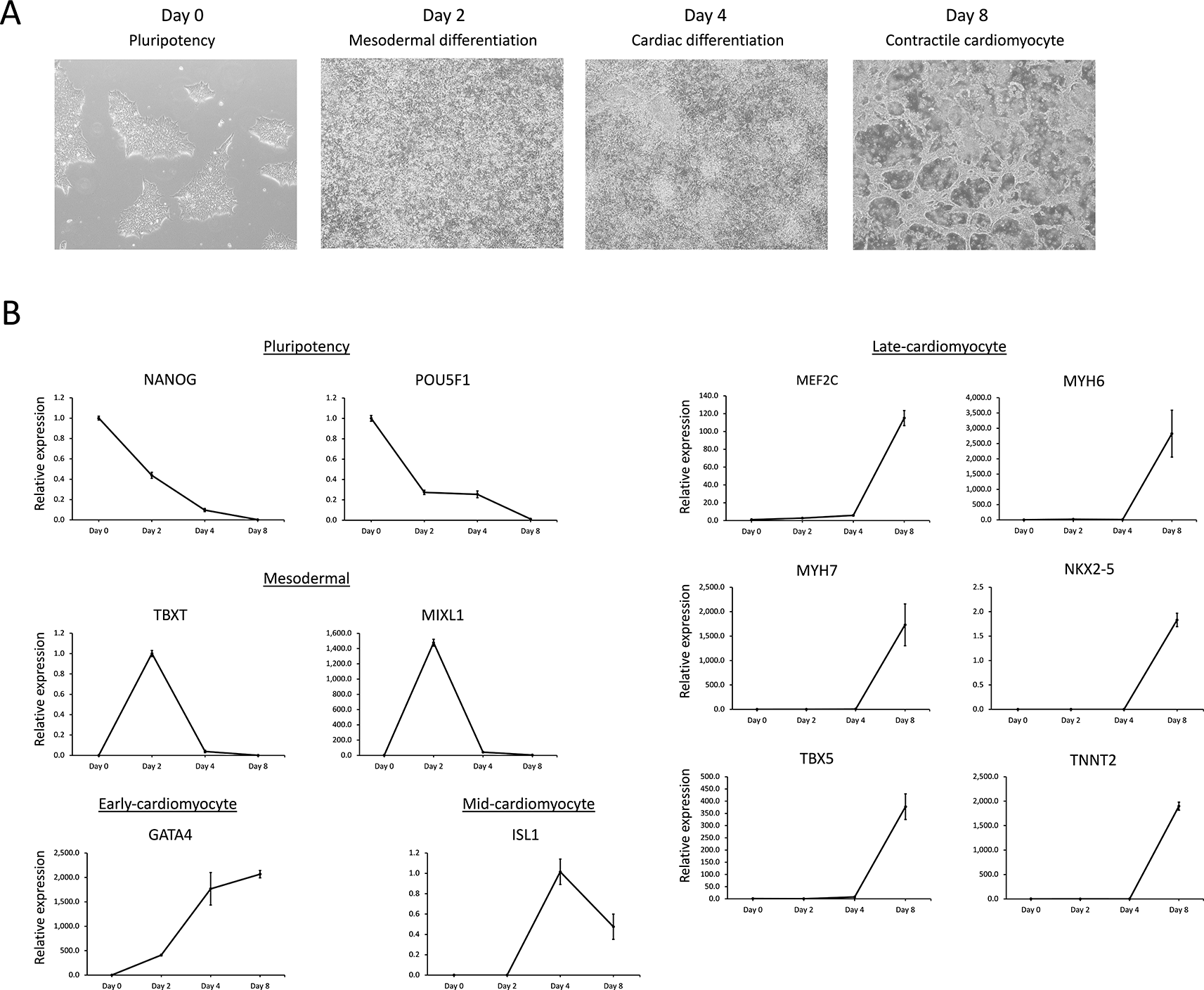
A) Micrographs of morphological changes in hiPSCs during cardiomyocyte differentiation. hiPSC colonies, characteristic of feeder-free human PSCs, were identifiable on day 0. Cell morphological changes are identifiable as differentiating cells proliferate to confluency by days 2 and 4, generating significant cellular debris. By day 8, the confluent multilayer cellular morphology of the well had undergone remodeling into rhythmically contractile cardiomyocyte syncytia.
B) Relative gene expression profiling using RT-qPCR of key cardiomyocyte differentiation markers obtained from the basic gene expression signature analysis for pluripotency and mesodermal, early-cardiomyocyte, mid-cardiomyocyte, and late-cardiomyocyte signatures within the GSE85331 (Liu et al., 2017) dataset. Note that ISL1 is the only mid-cardiomyocyte expression signature, whereas MEF2C, NKX2-5, and TBX5 exhibit late-cardiomyocyte expression.
To begin the in silico analysis of directed cardiomyocyte differentiation, we identified two publicly available GEO datasets that profile human cardiomyocyte differentiation (Figure 1). Both Refs. 17 and 18 utilize Illumina RNA-seq technology to profile gene expression changes during hPSC-directed cardiomyocyte differentiation. Although these reports utilized slightly different modifications to the original procedure, similar results were obtained with the robust and efficient differentiation of cardiomyocytes.11,13,17,18 Both datasets evaluated the major differentiation steps that were initiated by the addition of GSK3 inhibitors (day 0), the addition of WNT inhibitors (day 2 or 3), the addition of media supplemented with B27 and insulin (day 4 or 5), and then beating cardiomyocyte syncytia at the end of differentiation (day 15 or 30). Furthermore, these high-quality datasets evaluate cardiomyocyte differentiation using multiple hPSC lines, which allows the identification of cardiac developmental genes demonstrating consistent regulation and the exclusion of genes that exhibit variable expression between cell lines. Note that the bioinformatics analysis within these papers used specialized scripts in R and other languages to conduct their experimental analyses. The GEO-provided analysis software GEO2R can be used to define sample groups for analysis, set P value and fold change cutoff thresholds, select pairs of sample groups for differential expression analysis, and allow minimal plot functionality, data export, and the ability to download an R script that can be run locally. devGEA provides enhanced functionality for developmental time-series analysis in contrast to GEO2R, which will be explored in this report.
The human induced pluripotent stem cell (hiPSC) GibcoTM Episomal line (A18945) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The hiPSC line was derived from cord blood CD34+ progenitors using seven episomally expressed factors (Oct4, Sox2, Klf4, Myc, Nanog, Lin28, and SV40 T) and adapted to feeder-free, serum-free conditions. This line is indistinguishable from human embryonic stem cells and can be robustly differentiated into cardiac lineages.12
hiPSCs were maintained according to the manufacturer’s recommendations. Cells were cultured on Vitronectin (VTN-N) Recombinant Human Protein, Truncated-coated 6-well tissue culture plates (A14700, Thermo Fisher Scientific) using Gibco StemFlex™ Medium (A1517001, Thermo Fisher Scientific) maintained under normoxic conditions in 5.0% CO2. hiPSCs were maintained and renewed and passaged at least twice per weekusing Versene clump dissociation reagent(15040, Thermo Fisher Scientific) at multiple split ratios. The weekend-free Stemflex media passaging protocol was completed as described below. On day 1 cell passaging occurred as described below. VTN-N-coated 6-well plate were prepared by diluting 60-μL of the VTN stock solution (0.5 mg/mL) in 6 mL 1X Dulbecco’s Phosphate-Buffered Saline (DPBS) without calcium or magnesium (Thermo Fisher Scientific, 14190) to a final concentration of 0.5 μg/mL, adding 1 mL of diluted solution to each well, incubating the plate at room temperature for one hour, aspirating the solution, and then adding 1 mL of StemFlex Medium. Clump dissociation was performed by rinsing each well with 2 mL of DPBS, adding 1 mL of 1X Versene clump dissociation reagent(15040, Thermo Fisher Scientific) for 3-5 mins at room temperature, aspirating the Versene solution, and then removing cell clumps from the surface by gentle pipetting in 2 mL StemFlex Medium and transfer to a sterile 15 mL conical tube. The split ratios varied from 1:4 to 1:10 (0.5 to 0.2 mL) depending on the confluency and density of the hPSC colonies. The appropriate cell suspension volume was transferred to a new 15 mL conical tube, Stemflex media added to a final volume of 1 mL, mixed gently by pipetting, and then transferred into a well containing 1 mL of media. On day 2, wells were renewed with 2 mLs of Stemflex medium. On day 4, cells were Versene clump dissociated, passaged as described above. On day 5, cells were renewed with 4 mL StemFlex Medium. On day 8, cells were then passaged as described above.
Aliquots of cells were cryopreserved using the Gibco PSC Cryopreservation Kit (A2644601, Thermo Fisher Scientific) and recovered in stemflex media supplemented with 1X RevitaCellTM supplement (A2644501, Thermo Fisher Scientific) according to the manufacturer’s recommendations. For cryopreservation, an ~80% confluency well containing hiPSCs were Versene clump dissociated as described above except the entire cell suspension was centrifuged at 200 × g for 4 mins using an Eppendorf Centrifuge 5810 (Hamburg, Germany) followed by media aspiration. 2 mL of chilled PSC Cryomedia was then added to the cell pellet dropwise, gently mixed with 1 mL of cryo suspension aliquoted to cryovials (430488, CorningTM, New York, New York). Vials were then transferred to a Mr. Frosty™ freezing container and slowly frozen overnight at -80°C and then cryopreserved in the vapor phase of liquid nitrogen for long-term storage. For cryopreserved hiPSCs recovery, a vial was floated in a 37°C water bath until thawed and then transferred into a 15-mL conical tube. 10 mL of Stemflex media was added dropwise and then centrifuged. The cell pellet was then resuspended in 1 mL of Stemflex media supplemented 1X RevitaCell and transferred to one VTN-N-coated well of a 6-well plate containing 1 mL of Stemflex media supplemented with 1X RevitaCell. The next morning the well was renewed with 2 mL of Stemflex media and cultured as described above. Cell imaging was performed using an Olympus CKX41 Inverted Phase Contrast Microscope with a DP23M digital camera system and CellSens software (Evident, Tokyo, Japan). Micrographs were collected at 100× using the automatic settings.
Gibco Episomal hiPSCs were differentiated using the Gibco PSC Cardiomyocyte Differentiation Kit (A29212-01, Thermo Fisher Scientific) according to the manufacturer’s recommendations. Briefly, 70-85% confluent hiPSC cultures were dissociated into single cells usingTrypLETM Select Enzyme reagent (12563029, ThermoFisher Scientific), triturated, and resuspended in Stemflex media supplemented with 1X RevitaCell. Each well of a 6-well plate containing hiPSCs is rinsed with 2 mL of DPBS, 1 mL of TrypLETM Select Enzyme reagent (12563029, ThermoFisher Scientific) solution is added to each plate, incubated at 37°C for approximately 5 minutes until cells have detached from the well, and inactivated by adding 1 mL of Stemflex media. Cell suspensions are combined into a single 50-mL conical tube, pelleted by centrifuged at 100 × g for 5 minutes, and resuspended with 6 mL of Stemflex media supplemented with 1X RevitaCell. A 500 μL aliquot of cell suspension was removed to determine live cell counts and cell viability using the Bio-RAD TC20 automated cell counter and cell counting kit with trypan blue (Hercules, CA, USA). 20 μL of this suspension was combined with 20 μL of trypan blue solution with 10 μL transferred into one well of the counting slide. All cell counts were completed in duplicate. A cell suspension of 6 × 104 cells/mL was prepared in Stemflex media supplemented with 1X RevitaCell. One mL of cell suspension per well (6 × 104 cells/well) was plated onto a 12-well tissue plate coated with GeltrexTM LDEV-Free, hESC-Qualified, Reduced Growth Factor Basement Membrane Matrix (A14133, Thermo Fisher Scientific) after aspiration of the Geltrex solution. Geltrex-coated plates were prepared according to manufacturer’s recommendation. First, the concentrated Geltrex stock solution (15 mg/mL) was diluted in an equal volume of Dulbecco’s Modified Eagle Medium (DMEM) (11965092, Thermo Fisher Scientific) on ice to prevent gelling. This 180 μL of this solution was then added to a cold 15 mL conical containing 9 mL of DMEM on ice and mixed gently. 750 μL of this solution was added to each well of a 12-well plate and incubated at 37°C for 1 hour prior to aspiration and plating of the cells.
Twelve well plates were renewed with 1 mL of StemFlex medium each day until they density approached 60-80% confluency. Upon reaching the appropriate density (day 0), the wells were renewed with 1 mL of Cardiomyocyte Differentiation Medium A incubated for two days. On day 2, the wells were renewed with 1mL of Cardiomyocyte Differentiation Medium B. On days 6 and 8, the wells were renewed with 1 mL of Cardiomyocyte Maintenance Medium every two days. Small areas of beating cardiomyocytes were observed on day 6. Two days later (day 8), the cultures appeared to have undergone extensive remodeling.
Total RNA was isolated on the day of cardiomyocyte differentiation (day 0) and subsequently on days 2, 4, and 8. For each cardiomyocyte differentiation time point, three separate wells were harvested so that downstream gene expression analysis could be performed in triplicate. Cells were directly lysed by adding 0.3 mL TRIzol RNA isolation reagent (15596016, Thermo Fisher Scientific) to each well, vigorously rocking the plate for 5 minutes, and then freezing the lysate at -80.0 °C. Total RNA was isolated using the Direct-ZolTM RNA MicroPrep Kit (R2061, Zymo Research, Irvine, CA) according to the manufacturer’s recommendations, which included in-column DNase I treatment to remove genomic DNA. Total RNA was eluted in 10 μL of RNase/DNase-free water. One microliter was used to quantify the total RNA concentration and quality using a ThermoFisher Scientific NanoDrop™ One Microvolume UV-Vis Spectrophotometer. cDNA was prepared using the SuperScriptTM IV VILOTM Master Mix with ezDNaseTM Kit (11756050, Thermo Fisher Scientific) using 0.2 μg of total RNA according to the manufacturer’s recommendations, including ezDNaseTM pre-treatment to remove genomic DNA. cDNA synthesis reactions (20 μL) were diluted 1:10 to a final volume of 200 μL using RNase/DNase-free water to obtain a concentration of 1 ng/μL total RNA.
RT-qPCR was used to quantify the relative transcript expression between cardiomyocyte differentiation time points. Oligonucleotide primers were designed using Primer3 with standard parameters and amplicon product sizes ranging from 75 to 300 nucleotides, ensuring that each primer spanned at least one intron to ensure cDNA-specific application, and were produced by Integrated DNA Technologies using standard synthesis procedures (IDT, Coralville, IA).19 Table 1 lists the design and sequence information of each primer set. qPCR was performed using the PowerUpTM SYBRTM Green Master Mix (100029283, Thermo Fisher Scientific) system in an Applied BiosystemsTM QuantStudioTM 3 Real-Time PCR System (Thermo Fisher Scientific). qPCR reactions (10 μL qPCR reactions containing 2 μL (2 ng) of the reverse-transcribed total RNA, 1 pmol each of the relevant forward and reverse primers, and 5 μL of the 2X master mix PowerUpTM SYBRTM Green Master Mix (100029283, Thermo Fisher Scientific). qPCR reactions were performed in technical triplicates to evaluate the reproducibility of the quantification cycle (Cq) for each amplified condition. QuantStudioTM Design and Analysis Software VERSION 1.4.3 (Thermo Fisher Scientific) was used to run the experimental method, analyze, and determine the Cq values. This software is downloadable from https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/quantstudio-3-5-real-time-pcr-systems.html. The manufacturer’s standard cycling method consisted of a UDG activation step at 50.0 °C for 2 min, DNA polymerase activation step at 95.0 °C for 2 min, PCR amplification stage (40 cycles × 95.0 °C for 15 s, 58.0 °C, 72.0 °C for 1 min), and a dissociation curve stage (60.0–95.0° C at 0.1 °C/s) was used for all qPCR reactions. The qPCR experimental parameters utilized the standard curve experimental type setting, which determined the Cq value using both Auto-Threshold and Auto-Baseline analysis functions. The qPCR results were exported to MicrosoftTM ExcelTM for relative gene expression quantification. Relative gene expression was calculated using the 2-ΔΔCT method as described previously.20 The average standard deviation for all samples for the qPCR triplicate reactions was 0.159 Cq; the range between triplicate reactions for each sample varied from 0.003 to 0.544, demonstrating reproducibility near a standard deviation of 0.5 Cq. At least three No Template Controls (NTC) qPCR for each primer set were included in each experiment, validating the absence of template-independent amplification or template contamination. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene to normalize targets because it demonstrated a low Cq variance of 20.19 ± 0.73 (Mean ±).20,21
This table provides gene information, oligonucleotide sequences, and amplicon lengths for each gene-specific RT-qPCR experiment.
Figures 1 and 2 describe the small-molecule-directed cardiomyocyte differentiation used in these experiments11,13 our own study. Here, we highlight the key genes involved in developing our list of key marker genes and their expression patterns. First, the procedure involves seeding, recovering, and growing hiPSCs dissociated into single cells in pluripotency culture media.
The initial time point (day 0) of these cells in pluripotency culture media prior to subsequent media changes represents proliferative hiPSCs expressing the expected pluripotency markers POU Class 5 Homeobox 1 (POU5F1, also known as OCT4) and Nanog Homeobox (NANOG).22–24 On day 0, hiPSC media was replaced with cardiomyocyte differentiation media containing a GSK3 inhibitor, such as CHIR99021, which activates the canonical WNT signaling pathway and initiates mesodermal specification, as identified by T-Box Transcription Factor T (TBXT, also known as BRACHYURY [T]) and the early cardiac marker GATA Binding Protein 4 (GATA4).25,26 GATA4 is necessary for mesoendodermal development during gastrulation and is then expressed within the mesoderm and later in the cardiac lineage, in which it directs cardiac development and postnatal cardiac function.27–29 The divergence of day 2 induction of TBXT and GATA4 gene expression signatures enabled the classification of transient mesodermal expression from early cardiomyocyte gene signatures because GATA4 is robustly activated during mesodermal differentiation and then persists throughout cardiomyocyte differentiation (Figures 1B and 2B). Therefore, TBXT expression identifies transient mesodermal expression describing the mesodermal gene signature, whereas GATA4 represents the earliest state of cardiomyocyte gene induction, describing the early cardiomyocyte gene signature (Figure 1).
After 48-72 hours of incubation (days 2 or 3), the GSK3 inhibitor-containing media was replaced with media containing canonical WNT signaling inhibitors, such as IWP2 or IWP4 (Figure 1B). Canonical WNT inhibition induces the expression of several important progenitor genes that mark the cardiac lineages of early embryos.12,13 The cardiac progenitor markers ISL LIM Homeobox 1 (ISL1), Myocyte Enhancer Factor 2C (MEF2C), NK2 Homeobox 5 (NKX2-5), and T-Box Transcription Factor 5 (TBX5) exhibit unique expression profiles during later cardiomyocyte differentiation stages (Figure 1 and 2).30–33 Although these four genes are robustly expressed at high levels at the end of differentiation (days 8, 15, or 30, see Figure 1), the initial detection of transcripts occurs earlier in differentiation at lower levels.12,13 For example, while ISL1 transcripts in the GSE85331 (Liu et al., 2017) experiment demonstrated the greatest change in expression from day 2 to day 4, NKX2-5, MEF2C, and TBX5 genes were not maximally expressed until day 30 (Figure 6). A similar result was identified via RT-qPCR analysis of Gibco hiPSC cardiomyocyte differentiation (Figure 2B). Therefore, these progenitor markers represent two different gene expression signatures: a mid-cardiomyocyte gene expression signature with a maximum change in expression at day 4 (ISL1) or a late-cardiomyocyte gene expression signature on day 8 (MEF2C, NKX2-5, and TBX5) (Figures 1 and 2).
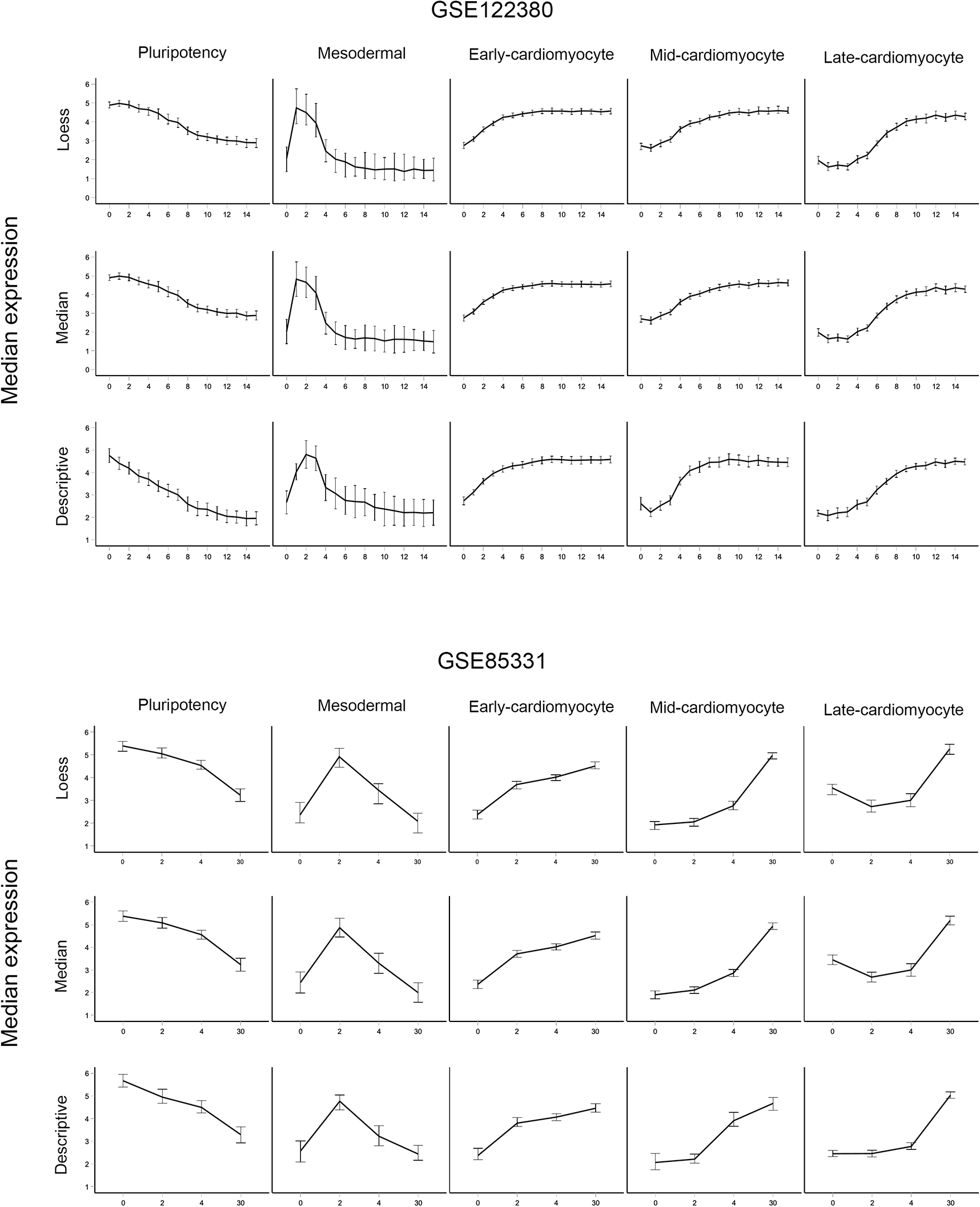
Median gene expression patterns of the five cardiomyocyte gene expression signatures were determined by LOESS, median, and basic analysis for GSE122380 (Strober et al., 2019) and GSE85331 (Liu et al., 2017). Each graph plots the median expression values and the 95% confidence interval error bars for pluripotency, mesodermal, early-cardiomyocyte, mid-cardiomyocyte, and late-cardiomyocyte-grouped genes.
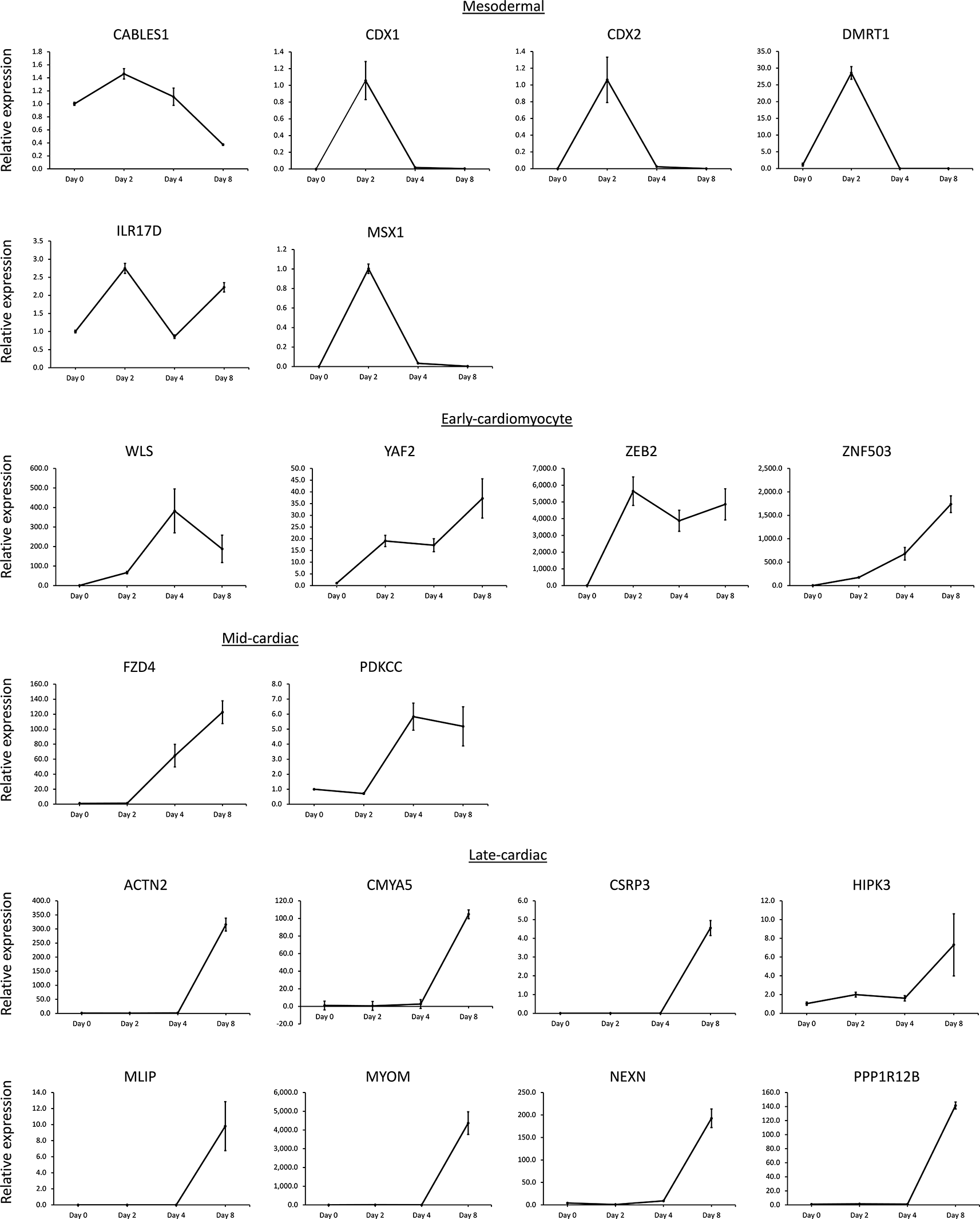
RT-qPCR analysis of candidate gene expression signatures during directed cardiomyocyte differentiation in Gibco hiPSCs. The plots are from the present study, with samples taken on Days 0, 2, 4, and 8. The grouping of genes/plots was based on the basic/descriptive gene analysis of the GSE85331 dataset (i.e., CABLES1 is listed under mesodermal because it was identified using the basic/descriptive analysis of GSE85331). RT-qPCR profiling approximates the expected gene expression signature for each stage of differentiation in this single analysis. However, discrepancies in these signature calls exist when these results are compared to those of some of the classification methods discussed in the article.
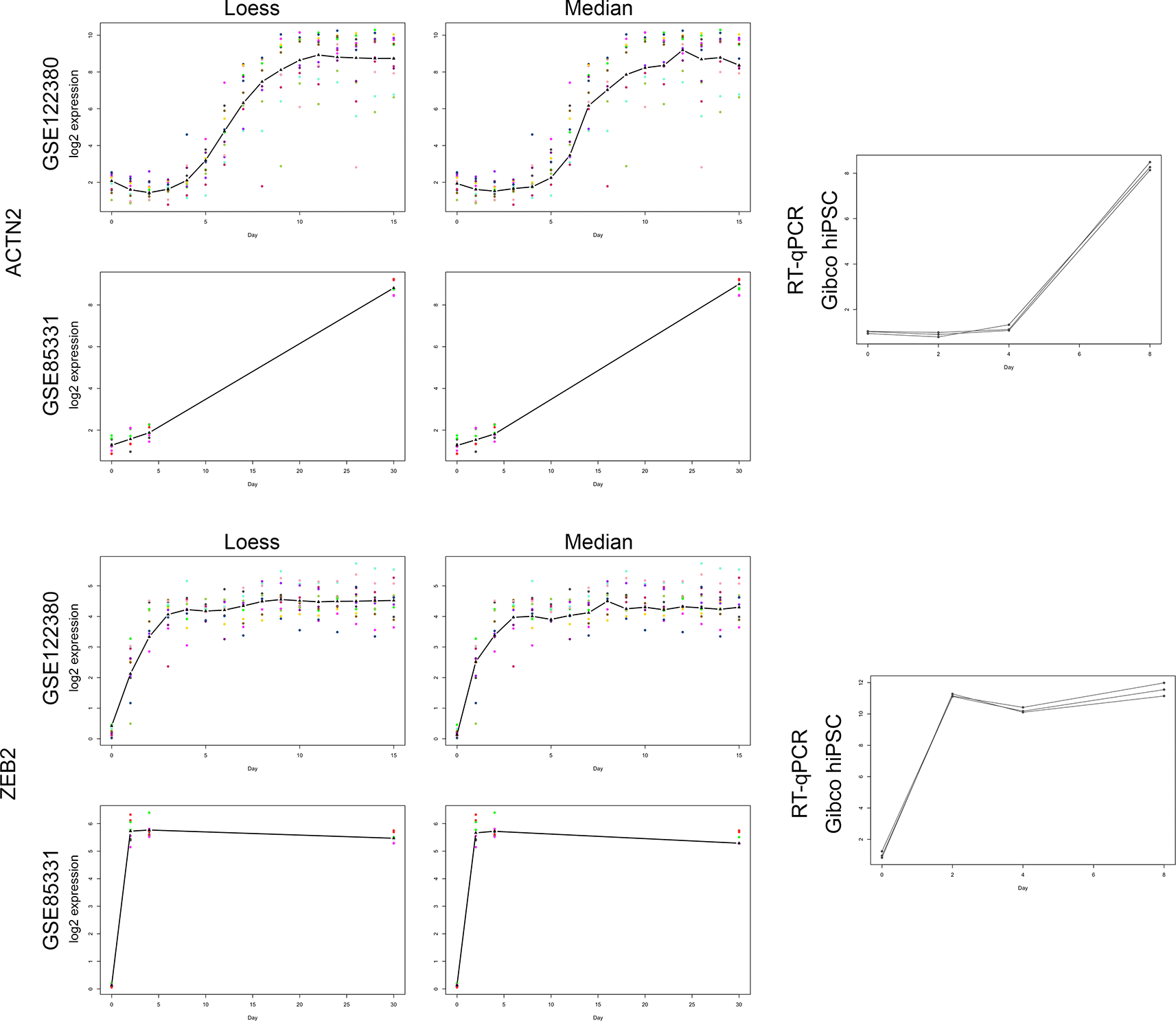
Comparison of correlation curve fitting models for GSE122380 and GSE85331 to RT-qPCR analysis of ACTN2 (a late-cardiomyocyte signature call) and ZEB2 (an early cardiomyocyte signature call). These two genes have been used as examples in the text to describe our classification methods. To evaluate the LOESS (left panel) and median curve-fitting correlation methods (middle panel), the log2 expression values for each time point were plotted for both datasets, and the line overlay identified the respective curve-fitting model. The additional time points located within GSE122380 (16 time points) enhanced the gene expression curve compared with GSE85331 (four time points). Although both methods generated similar curves for each gene, the LOESS function provided additional smoothing to the plot. The right panel shows log2 relative gene expression changes during cardiomyocyte differentiation of GIBCO hiPSCs using RT-qPCR. Note the gene expression signature similarities among the three analytical methods.
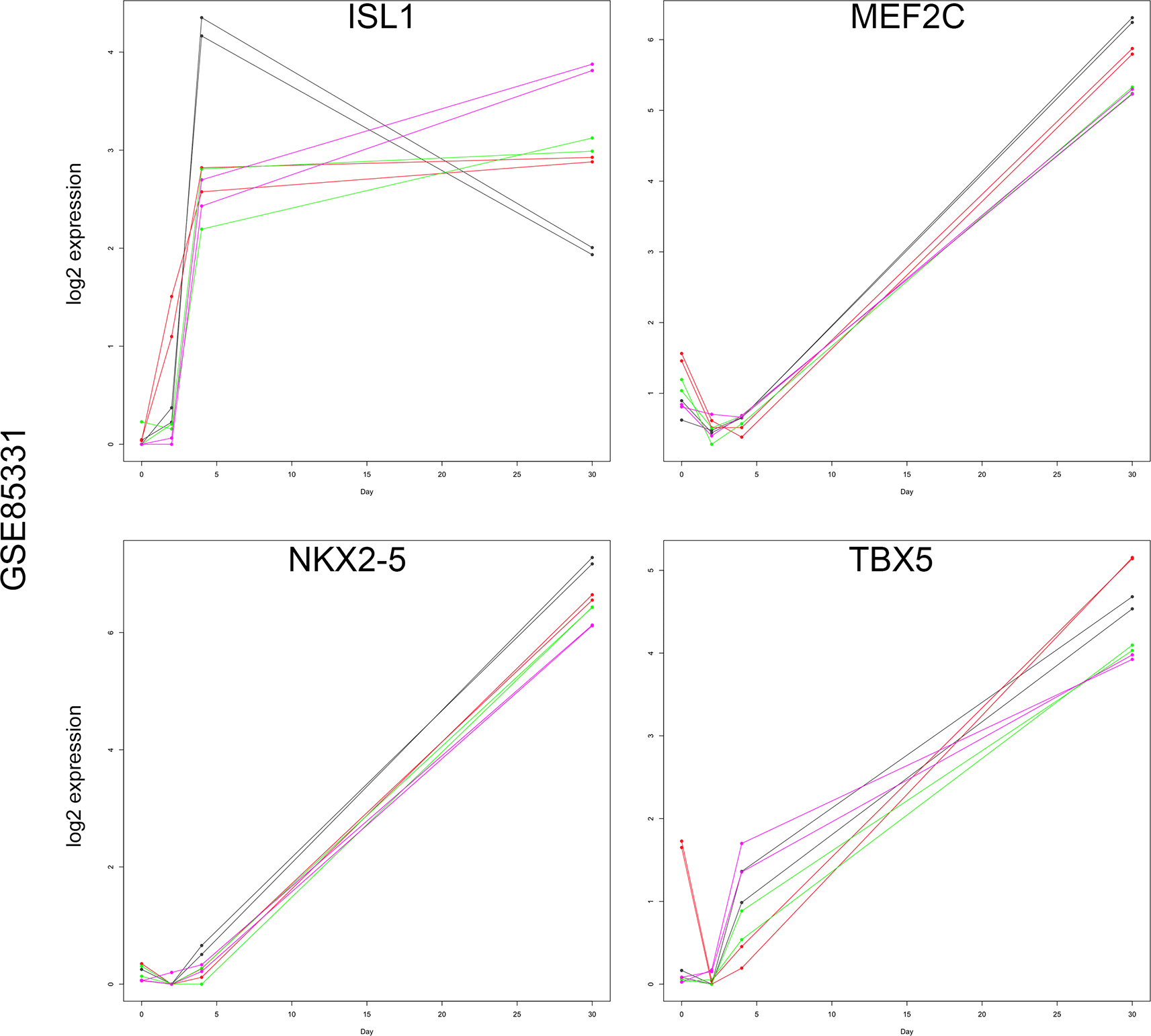
Graphs of day 0, 2, 4, and 30 log2 expression values for ISL1, MEF2C, NKX2-5, and TBX5. Each line represents an individual cell line and replicates. Note that ISL1 and TBX5 demonstrated marked expression at day 4, whereas MEF2C and NXK2-5 exhibited baseline expression until day 30. Therefore, ISL1 and TBX5 were the best candidates for representing the mid-cardiomyocyte gene expression signature.
After 48 h of cardiomyocyte progenitor induction (day 4-5), a cardiomyocyte-maintenance medium containing B-27 supplement and insulin was added, which supports cardiomyocyte differentiation and viability. Subsequently, key contractile cardiomyocyte markers, such as MYH6, MYH7, and TNNT2, are highly expressed.24,34 After the onset of contraction on approximately day 6-7 (Figures 1 and 2), the differentiated cardiomyocytes underwent remodeling into syncytia, with rhythmic contraction apparent at the gross level (Figure 2A). The expression patterns of the genes described above were utilized as key cardiac gene expression signatures for the classification of differentially expressed genes during cardiomyocyte differentiation in the preceding experiments (Figures 1 and 2).
Here, we describe the main features and workflow of devGEA. We look at these features in more detail in the Results and Discussion section where we use the software to analyze the prior datasets (Refs. 17 and 18).
devGEA is a shiny web application written completely in the R programming language. We also provide a self-contained R markdown file with the analysis that is contained in the Results and Discussion section.
Data import, normalization, and plots. The devGEA shiny web application can import data as raw counts or already normalized. Data files can be imported by uploading a file or specifying a GEO ID. The web application allows for normalization using limma. Data can be scaled by taking the logarithm of the data or scaling it to have a mean of 0 and standard deviation of 1. Data can be filtered based on a raw count threshold and whether the gene information is missing. The data and results can be viewed in an interactive table format so that the user can verify the data import, analytical methods, and results. The table can be searched using gene symbols or identifiers, or by setting ranges for columns. Gene selection by mouse clicks provides a basic gene expression plot.
Additional metadata for the samples (e.g., time point, cell line) can be specified by importing a csv/tsv file, using a regular expression based on the column headings, or manually entered within the app. Once the sample metadata have been imported, the sample groupings used for analysis are specified (e.g., groups based on time point or cell line). Additional metadata for genes (e.g., gene symbols or names) can be imported automatically from Ensembl BioMart35 through the app.
Additional data normalization plots were provided to evaluate the quality of the dataset. Gene expression box plots or histograms for each sample were used to identify outliers. The PCA of the samples or groups provides a linear representation of variation within the data set. Samples identified as outliers or with poor gene expression preparation can then be marked for exclusion, and the data can be reanalyzed.
Analysis. Once a dataset is loaded, the user can choose to add an analysis. For a basic analysis, the software computes for each gene – medians of each time point, maximum and minimum time points based on expression, and other simple descriptive statistics of the data. These results are displayed within a table and can be exported; results can be filtered based on fold change, maximum/minimum time point, or time point of maximum change. For a correlation analysis, the user specifies either a set of marker genes or a set of gene expression patterns. For each gene in the dataset, the software computes the correlation of the gene with each marker gene (or given gene expression pattern) and then classifies each gene based on the highest correlation. The results of this analysis are the correlations and classifications for each of the genes. We discuss this analysis technique further in the Results and Discussion section. For an ANOVA analysis, the user specifies a sample property (e.g., time point or sample group) to define a linear model, and the software computes adjusted p values for each gene based on this property. The results of the analysis are the adjusted p values for each gene. For a subset analysis, the software combines results from previous analyses within the app (i.e., by taking intersections or unions of result sets). By default, the software takes the intersection of each successive analysis (i.e., first filtering based on fold change with basic analysis, and then filtering based on adjusted p values with ANOVA analysis, and so forth).
Saving and exporting results. The entire project can be saved as an R data file and then later loaded into the software again. Result sets can be exported in csv format. Plots can be exported as svg files.
We note that the main novelty in devGEA is the correlation analysis, which is a novel form of analysis in this setting (as far as we are aware). We have built the standard steps of differential gene expression analysis around the correlation analysis so that users with little or no programming experience can use this form of analysis themselves.
devGEA is written completely within the R programming. The code has been tested on macOS 13.4, Windows 10, and Ubuntu Linux.
The shiny web application can be downloaded and run locally or can be run from our server. To run locally, the user runs the file app. R that is within the shiny_app directory of our gitlab repository (https://gitlab.com/jkinne/devgea).
When the shiny web application is run from our server (https://cs.indstate.edu/devgea), the user does not require to install any software on their own system. We note that we use ShinyProxy36 as the backend on our server; we provide example configuration files within the shinyproxy directory of our gitlab repository.
To run the R markdown file that contains the analysis in this paper, the user should knit the file devGEA. Rmd that is in the gitlab repository.
Here, we describe our analysis of the prior datasets (Refs. 17 and 18). We focus our discussion on the analysis provided within the Rmd. After describing our analysis, we discuss validation with our study results.
Many researchers argue for using transcripts per kilobase million (TPM) as the preferred normalization for RNA seq count data (see Ref. 37 which introduced the term, and more recently38 which evaluated different normalization methods). The National Center for Biotechnology Information (NCBI) has recently begun to provide both raw counts and normalized files for all GEO datasets. We used the TPM-normalized data available from NCBI as the input for our analysis. The NCBI database also provides supporting gene annotations.
We filtered the data so that we only considered protein-coding genes, and we also only considered genes that were non-zero in at least two samples and had a total TPM sum of at least 10.
We extracted metadata about the samples using series matrix files that can be downloaded from the NCBI GEO. The design matrix contains the following for each sample: cell line, time point (day #), replicate #, sample ID, whether to exclude the sample from analysis, and sample name for use on plots.
As is standard for biological data, we take the logarithm (base 2) and use these values for further analysis.
Evaluating cell lines - Ref. 18 used a Gaussian process model to identify clusters of cell lines that share gene clusters. One of the cell line clusters was noted to have less robust expression of cardiac troponin. Thus, we removed these cell lines from the analysis. Many approaches can be used to cluster cell lines and identify outliers. We considered an approach similar to Ref. 39, but decided to stick with the clustering of cell lines identified in Ref. 18.
Combined analysis - We considered whether the two datasets could be combined and analyzed together.40 lists a number of potential problems when attempting to combine datasets from separate experiments or laboratories. In this case, it was determined that the datasets should not be combined. As can be seen from the principal components analysis (PCA) plot in Figure 7, the samples from the datasets are well separated on the PCA plot. Notably, Refs. 18 and 17 used different laboratory methods to filter out ribosomal RNA -17 used rRNA depletion, while18 used poly(A)+ selection. This is one of the potential inconsistencies that Ref. 40 warns.
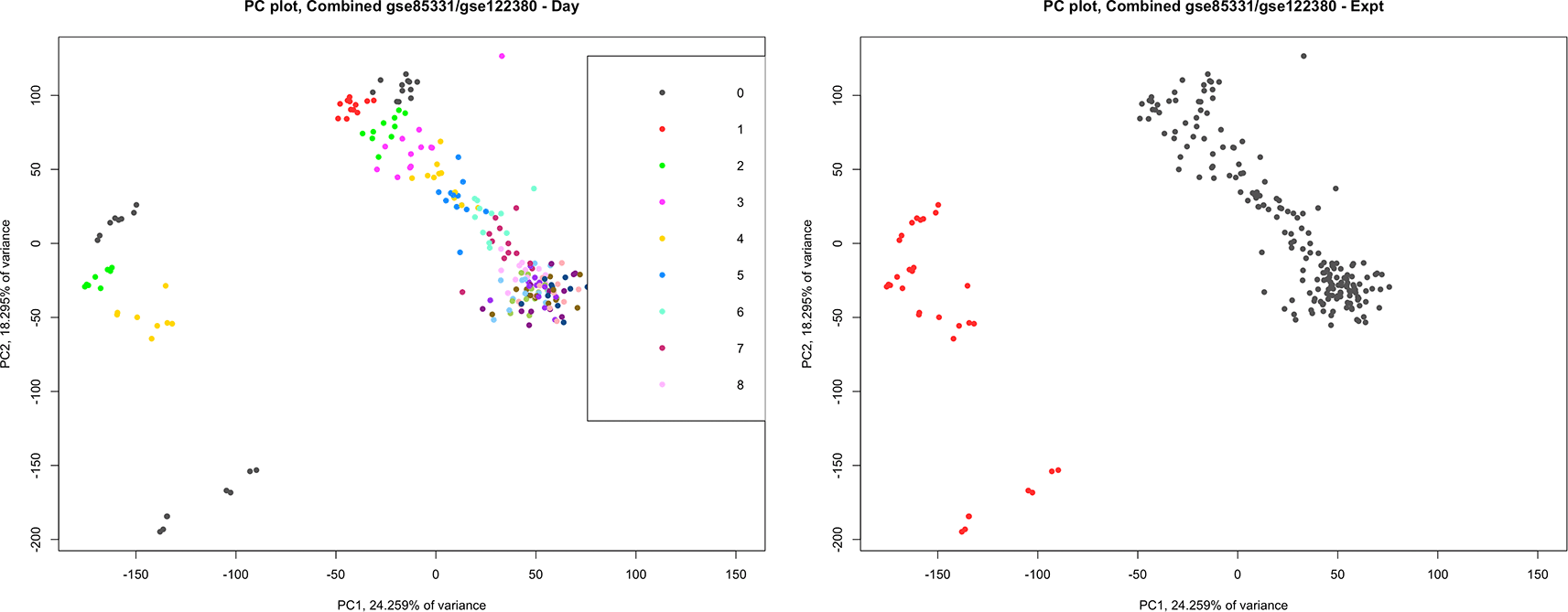
A principal component analysis plot combining the datasets GSE122380 and GSE85331. We considered whether the two datasets could be combined in a single analysis. On the left, each sample is colored by day. Note that the samples are clustered by day, going from top to bottom and left to right. On the right, each sample is colored according to the experiment, and we see that GSE122380 and GSE85331 are clearly separated on the PCA plot.
We identify and cluster significant genes using a variety of methods.
• Linear model ANOVA was performed using a linear model over the key time points to obtain adjusted p-values.
• Basic/descriptive statistical analysis - using simple conditions related to the highest time points of expression and the largest change in expression.
• Correlation analysis using the correlation of each gene with key marker genes or expression patterns of interest.
We suggest that our basic analysis and correlation analysis as types of analysis should be considered when known patterns of interest or marker genes are known. Our precise method of using correlation to cluster is novel (to our knowledge at the time of writing), although it is well known that using correlation is preferred for computing distances with biological data (see e.g., Ref. 41). These methods are discussed in detail in the following sections.
Some parts of our analysis looked at gene expression at only the key time points as described earlier: days 0, 2, 4, and 30 in Ref. 17 and days 0, 3, 5, and 15 in Ref. 18. Other parts of the analysis are time-series methods that use all the available time points.
For each gene, we used a linear model over the four key time points. The resulting adjusted p-values were used to indicate if a gene had a statistically significant difference between at least two of the key time points. This is the standard step in the analysis process.
Differential gene expression analysis described above with standard cutoff for adjusted p value of 0.05 and log2 fold change of at least 1 identified 8634 genes within the GSE85331 dataset and 7117 genes within the GSE122380 dataset that had a significant difference among the four key time points, with a total of 5241 genes common between both lists. We will further filter by requiring that each gene has a correlation of at least 0.75, one of our key marker genes (POU5F1 (pluripotency), TBXT (mesodermal), GATA4 (early-cardiomyocyte), TBX5 (mid-cardiomyocyte), and TNNT2 (late-cardiomyocyte)), using the correlation analysis that is described later. This provided a list of 3259 genes for further investigation.
Table 2 and Figure 5 present the main analysis results for two example genes: ZEB2 and ACTN2. As indicated in the table, the adjusted p-values for both genes were 0 for both datasets. Both genes also passed the log2 fold change criteria for both datasets, and had a correlation of at least 0.75 for their closest matches, for the correlation analysis. Therefore, both genes were in the 3259 gene set.
A summary of the analysis of the two genes is provided.
The expected classification for ZEB2 is early, and each analysis method (in bold) classifies ZEB2 as early. The expected classification for ACTN2 is late, but some classification methods classify ACTN2 as mid-cardiac.
The class (descriptive) is based on the time points of the maximum expression and the time point at which there is a maximum change. For gse122380, the major time points used for descriptive analysis were days 0, 3, 5, and 15; for gse85331, the major time points were days 0, 2, 4, and 30. ZEB2 is classified as early because it has the maximum change going into the second major time point and its maximum expression in one of the last two time points. ACTN2 is classified as late because it has the maximum change at the last major time point and has the maximum expression at the last major time point.
The loess class was based on the correlation of the loess fit of the gene with the loess fits of the marker genes for each class (pluri - POU5F1, meso-TBXT, early - GATA4, mid - TBX5, late - TNNT2). The median class is based on the correlation of the day median of the gene with the day median of the marker genes. ZEB2 was classified as early in each of these methods with both datasets because its correlation was closer to GATA4 than to the other marker genes. For example, for gse122380 and the loess fit, the loess fit of ZEB2 was 0.96 with GATA4, and the second highest correlation was with the mid-cardiac marker gene TBX5 (0.75). ACTN2 is classified as late according to the loess and day median fit for gse122380, but it is classified as mid-cardiac for both fits for gse85331. For example, for gse85331 and the median fit, ACTN2 had a correlation of 0.99 with the mid-cardiac gene GATA4, while it had a correlation of 0.95 with the late marker gene TNNT2. Note that ACTN2 has a high correlation with both, and this is also the case for many genes that are classified as either mid- or late-cardiac.
Small-molecule cardiomyocyte differentiation is an easy and effective procedure involving three media changes that allows transcriptomic profiling throughout differentiation. Coinciding with each medium change, subsequent gene expression changes should proceed in a straightforward manner, representing the four major stages of cardiomyocyte differentiation. Therefore, the following rules can classify the major gene expression signatures according to the day of maximum expression (DayMax) and the day of maximum difference in the time series (DayMax-difference) (Figures 1 and 2).
• Pluripotency – DayMax occurring on day 0 similar to POU5F1 and NANOG expression.
• Mesodermal – DayMax at day 2/3 and maximum difference centered on day 2/3 (either positive DayMax-difference occurring on day 2/3, or negative DayMax-difference occurring on day 4/5), similar to TBXT and MIXL1 expression.
• Early-cardiomyocyte – DayMax occurred later in differentiation on days 4/5 or 8/15/30 since expression persisted throughout cardiomyocyte differentiation and a positive DayMax-difference on day 2/3, similar to GATA4 expression.
• Mid-cardiomyocyte – DayMax occurred on days 4/5 or 8/15/30 and a positive DayMax-difference was observed on day 4/5, similar to ISL1 expression.
• Late-cardiomyocyte – DayMax occurring on days 8/15/30 and positive DayMax-difference on day 8/15/30 gene, similar to TNNT2 expression.
Consider the example genes, ZEB2 and ACTN2. The middle plots in Figure 5 show the median values at each time point. For GSE85331, ZEB2 had its maximum value on day 4 and the maximum difference in expression on day 2. ZEB2 was thus classified as an early cardiomyocyte in GSE85331. For GSE122380, we analyzed a subset of time points: days 0, 3, 5, and 15. Among these time points, the maximum expression of ZEB2 was at day 15, and the maximum difference in expression increased from day 0 to day 3. ZEB2 was thus classified as an early cardiomyocyte for GSE122380. For ACTN2 in GSE85331, the maximum expression was observed at day 30, with the maximum difference in expression occurring at day 30. In GSE122380, considering only days 0, 3, 5, and 15, the maximum expression of ACTN2 was on day 15, with the maximum difference going up from day 5 to day 30. ACTN2 was classified as a late cardio for both datasets. Table 2 contains a summary of the analysis for ZEB2 and ACTN2, with the “class (descriptive)” given for each matching our discussion here (early for ZEB2 and late for ACTN2).
Classification using simple descriptive statistical rules is understandable. If a gene of interest is misclassified via user-defined rules, it is easy to determine the cause. In contrast, sophisticated unsupervised clustering methods may generate unexpected gene cluster patterns that require additional filtering or parameter modifications to generate results usable by the investigator. We propose that the simple basic analysis described here may aid investigators in data-exploration tasks.
The basic descriptive analysis of GSE85331 and GSE122380 datasets identified 1711 genes within the 3259 gene list that possessed identical signature calls for this basic/descriptive analysis for the POU5F1 (pluripotency), TBXT (mesodermal), GATA4 (early-cardiomyocyte), TBX5 (mid-cardiomyocyte), and TNNT2 (late-cardiomyocyte) classifications in this basic analysis. As indicated in Table 2, the example genes ZEB2 and ACTN2 are given the same “class (descriptive)” for both datasets (early for ZEB2 and late for ACTN2); therefore, both are on the 1711 gene list. The median expression patterns of each gene signature classification for descriptive analysis of both datasets are shown in Figure 3. These cardiac gene signatures approximate previous investigations of cardiomyocyte differentiation, demonstrating a robust method for precisely classifying gene expression patterns in a developmental system.12,13,15
devGEA shiny web application: The shiny web application computes the basic statistical properties of a dataset and allows the user to define clustering groups and rules for the clustering groups. The shiny web application can thus be used to easily perform the basic/descriptive analysis discussed here, as well as filtering based on p values and fold changes without requiring any programming or software installed locally.
An independent experimental analysis of cardiomyocyte differentiation using a commercially available hiPSC cell line was performed to evaluate the success of gene expression signature calls and to validate the candidate cardiac genes identified in this analysis. The episomal GIBCO hiPSC line was obtained, expanded, and differentiated into cardiomyocytes using commercially available reagents. As expected, hiPSC differentiation proceeded as expected: on day 0, independent hiPSC colonies were visible; from days 2 to 4, differentiating cells had proliferated to confluency with detaching and dead cells visible in the culture dish; on day 6, small contracting areas within the cellular lawn were identifiable and proceeded to undergo remodeling to large rhythmically contracting web-like syncytia (Figure 2A). At equivalent time points to the GSE85331 experiment, total RNA was extracted, and RT-qPCR was used to validate relative gene expression across the experiment. The relative expression of each gene was determined by the 2-ΔΔCT method using Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) expression for normalization. The set of 32 genes tested in this way are listed in Figures 2 and 3 (RT-qPCR results) and Table 3 (summary of our analysis of these genes in the prior datasets). We now describe the set of 32 genes, their RT-qPCR results, and the analysis of the prior datasets.
Summary of the analysis of the 32 genes for which we performed qPCR measurements. The results are given for each of the two datasets under which we analyzed the genes. First, the expected classification of the gene, gene id, and gene symbol are given. Then, for each dataset (gse122380, gse85311), the following are given: adjusted p values, log2 fold change between the highest and lowest major time point, classification by basic/descriptive analysis, classification by loess fit, correlation of loess fit to classified marker gene (pluri - POU5F1, meso-TBXT, early - GATA4, mid - TBX5, late - TNNT2), classification by day median correlation, and correlation of day median correlation classification.
POU5F1 and NANOG expression was identified to verify pluripotency and the subsequent downregulation of pluripotency factors during differentiation (Figures 1 and 2B). Ten key cardiomyocyte differentiation markers were selected for evaluation and compared with the 1711 gene descriptive analysis results (Figure 2B). All ten differentiation markers were identified in the 3259 descriptive analysis list, while all but three (ISL1, MEF2C, TBX5) were present in the 1711 gene list. While pluripotency and mesodermal and early cardiomyocyte expression patterns were comparable between the RT-qPCR and descriptive analysis results, several discrepancies were identified within the mid-cardiomyocyte and late-cardiomyocyte expression pattern groups. For example, ISL1 demonstrated robust activation at day 4 (a mid-cardiomyocyte signature) in the RT-qPCR data but was absent from the 1711 gene list, indicating variable expression between the two prior datasets. Interestingly, MEF2C and NKX2-5 levels determined via RT-qPCR were extremely low or undetectable until day 8, which is consistent with a late cardiomyocyte signature rather than a mid-cardiac signature (Figure 2B). Within the 3259 gene list, NKX2-5 was classified as a late-cardiomyocyte signature in both datasets, while the MEF2C classification was mismatched between mid- and late-cardiomyocyte signatures in the prior data sets. The mid-cardiac genes ISL1, MEF2C, and NKX2-5 are important cardiac regulatory genes necessary for cardiac lineage development, cardiomyocyte differentiation, and proliferation. This inconsistency may result from the delayed induction of key cardiac genetic regulatory genes during cardiomyocyte differentiation. Additionally, biological variation or experimental differences between hiPSC lines and/or experimental methods may be responsible for these discrepancies. Therefore, these expected mid-cardiomyocyte genes possess similar expression profiles to late-cardiomyocyte genes, such as sarcomeric TNNT2 which is highly expressed prior to contraction. Interestingly, NKX2-5 expression marks the cardiac mesoderm, which later gives rise to a linear heart tube in early mouse embryonic heart development.11,30,33,42 The later-than-expected expression of these genes may represent a lagging entrance of differentiating cells into the cardiac progenitor phase or alterations to the overall cardiogenic program.
Next, twenty genes with previously unvalidated gene expression patterns in hiPSC cardiomyocyte differentiation representing various cardiomyocyte differentiation signatures as called by the GSE85331 basic/descriptive analysis were selected (six mesodermal genes, four early-cardiomyocyte genes, two mid-cardiomyocyte genes, and eight late-cardiac genes) and investigated for reproducibility using RT-qPCR (Figure 3). RT-qPCR was performed on each gene to establish the gene expression signature pattern in the GIBCO hiPSC line. The RT-qPCR expression patterns for the 20 genes were classified according to the descriptive signature call in the GSE85331 dataset (Figure 3). RT-qPCR expression patterns approximately matched the descriptive signature classifications within the RNA-seq data sequence.
To increase the rigor of this analysis, the 32 RT-qPCR genes investigated were filtered, requiring descriptive signature calls to match the GSE85331 and GSE122380 analyses. 29 of the 32 genes possessed the same descriptive signature classification between the two data sets. Interestingly, CABLES, ISL1, MEF2C, and TBX5 did not meet these criteria, indicating variability between the datasets. The expected mid-cardiomyocyte genes, ISL1 and TBX5, were not successfully replicated in these datasets, indicating potential difficulty in distinguishing mid- and late-cardiomyocyte genes because of their similarity in gene expression, perhaps due to differences in the activation of the cardiomyocyte differentiation program. Although discrepancies exist between the datasets, this stringent approach demonstrates the advantages of utilizing the descriptive analysis method to identify gene expression patterns.
Whereas gene signature classification utilizing hierarchical clustering, k-means clustering, and self-organizing maps relies on classification techniques that randomly sample the dataset, we utilized a method that classifies genes based on correlation with user-specified gene or expression profiles that model the gene signature patterns of interest. This method enables the user to utilize a priori knowledge of gene expression or to explore gene expression patterns of interest based on their analyses. It also provides reproducibility between analyses and flexibility in selecting genes based on the extent of correlation with a gene model. In correlation analysis of cardiac signatures during differentiation, specific gene models can be selected within the dataset based on prior knowledge.
Key marker genes - The following results were obtained from using the following cardiac marker gene signatures represented by a specific cardiomyocyte differentiation gene: POU5F1 (pluripotency), TBXT (mesodermal), GATA4 (early-cardiomyocyte), TBX5 (mid-cardiomyocyte), and TNNT2 (late-cardiomyocyte). Although ISL1 is a potential mid-cardiomyocyte signature due to its expression during early cardiac progenitor development, ISL1 was not considered a gene signature because the signature calls using our basic/descriptive statistical analysis did not match between the GSE85331 and GSE122380data sets (and thus ISL1 is not on the 1711 gene list). Instead, we arbitrarily selected TBX5 as the mid-cardiomyocyte gene expression signature for the following correlation analyses because it demonstrated moderate upregulation on day 4 or 5 within the RNA-seq and RT-qPCR data sets compared to later upregulation of the late-cardiomyocyte marker TNNT2 after this time point (Figure 5). Within the RT-qPCR data, TBX5 relative expression is moderately increased from day 2 (0.45) to day 4 (7.68); however, this expression was upregulated nearly 50 times on day 8 (49.12), whereas TNNT2 demonstrated several magnitudes of upregulation from day 4 (0.88) to day 8 (1899.00).
LOESS and medians correlation: Two different methods were used to determine correlations between each gene and the cardiac marker genes within the 3259 priority cardiomyocyte gene list (see above). The LOESS smoothing technique uses a model to fit each gene to the cardiomyocyte differentiation marker gene and then computes the correlation between the fit of each gene and the fit of each cardiac marker gene. The second method computes a median value for each time point for each gene, and then correlates the results with the medians of each cardiac marker gene. After computing the correlations (using either LOESS fit or medians), each gene was classified according to the cardiac marker gene with the highest correlations. Each gene was correlated to a specific gene model, with the highest correlation selected for a cardiac expression signature.
devGEA shiny web application: We note that the shiny web application includes gene clustering based on median correlation but does not yet (at the time of writing) contain LOESS correlation clustering.
Example genes: Consider example genes ZEB2 and ACTN2. The LOESS fits are given in the plots on the left side of Figure 5, and the median values are given in the middle plots. Table 2 shows the correlation between these values and those of each of the key marker genes. For example, ZEB2’s LOESS model fit for GSE122380 was 0.966721808 with the model fit of the early marker gene (GATA4), with the second-highest correlation being 0.745102698 with the model fit of the mid-cardiac marker gene (TBX5). ACTN2 was assigned a different classification using the LOESS fit for the two datasets (late-cardiomyocytes for GSE122380 and mid-cardiomyocytes for GSE85331). For GSE85331 and LOESS fit, the LOESS fit for ACTN2 had a correlation of 0.987544522 with the mid-cardiomyocyte marker gene (TBX5), with the second highest correlation of 0.95867473 with the late-cardiomyocyte marker gene (TNNT2). Note that both mid- and late-cardiomyocyte signatures are reasonably good fits for ACTN2 in the correlation analysis, while the drop-off in correlation with the other marker genes is much larger.
Filtering: A minimum correlation value of 0.75 was selected as an arbitrary cut-off to further filter the differentially expressed genes (the LOESS fit and median values should have a correlation of at least 0.75, assigned to least one of the key marker genes). The mean correlation factor for this gene list was 0.931 with a standard deviation of 0.057, demonstrating the combined effectiveness of this filtering and classification strategy. The mean expression patterns of all genes in a particular signature classification reflected the corresponding model in both the datasets (Figure 4).
Comparisons: The comparison of the LOESS and median correlation methods within each dataset was surprisingly robust. 93% of the 3259 genes within the GSE85331 dataset were classified into the same signature classification (same classification using LOESS correlation and using median correlation), while 91% of the genes in the GSE122380 dataset were similarly classified using LOESS and median correlation. The comparison between GSE85331 and GSE122380 datasets was less robust. Using the LOESS method, 58% of the 3259 genes were successfully classified into the same signature using the LOESS method between the GSE85331 and GSE122380, while 59% of the genes were successfully classified using the median correlation method in the two data sets. Consider two example genes: ZEB2 and ACTN2. As indicated in Table 2, ZEB2 has the same correlation classification using both methods and both datasets, so ZEB2 is among the genes counted for all these percentages. ACTN2 has the same classification using LOESS and medians fit for GSE122380 (so is among the 91%) and has the same LOESS and median classifications for GSE85331 (so is among the 93%). ACTN2 has different classifications for both datasets for median correlation (not among the 59% mentioned) and has different classifications between the two datasets for LOESS correlation (not among the 58% mentioned).
The median gene expression signatures of each method and RNA-seq dataset are shown in Figure 4. Discrepancies may reflect a combination of biological and technical variations and analysis limitations due to differing numbers of time points between the RNA-seq experiments of the differentiation procedures. Further contrast of these analyses requiring both datasets to give the same classification for both methods (GSE122380 provides the same classification for both LOESS and medians, and GSE85331 provides the same classification for both LOESS and medians) identifies a total of 1704 of the 3259 gene lists reproducibly assigned to the same signature.
Next, we compared our RT-qPCR results with the LOESS and median correlation results of the inter-RNA-seq dataset comparisons. A summary of the analysis of these 32 genes is provided in Table 3. Of the 32 genes investigated, 27 RT-qPCR genes were identified in 3259 genes. Interestingly, 13 passed all filters and were classified the same using all six of our classification methods (basic/descriptive on both datasets, loess correlation on both datasets, and medians correlation on both datasets). Of these, TNNT2 was the only gene with an expected classification as mid or late. Most of the other mid/late genes were not classified consistently or, as expected, in GSE85331. This points to the difficulty in distinguishing the later time points in the experiment, although we expect that we can obtain a higher degree of matching by either selecting different key marker genes or using constructed gene patterns rather than the model fits of the marker genes. We report the results based on our initial plans to avoid simply selecting the parameters that yield the best results. Even so, most of the 32 genes in the mid/late phase that do not have 6-way agreement have high correlation values for both mid and late marker genes, meaning that we should consider not only the marker gene with the highest correlation, but also the second highest correlation and group.
Finally, we considered which of the 3259 genes were classified as the same using all six methods. Of the 3259 genes, 840 were classified as the same using all six methods.
Overall, this investigation demonstrates the unique analytical methods that devGEA provides to classify gene expression signatures in order to organize and study high-throughput developmental time-series gene expression studies. In addition, these studies suggest that caution must be taken when interpreting these analyses because of the inherent variability between experiments. Multiple methodological approaches may be necessary to evaluate the biological processes that are investigated to obtain the best representation of gene expression data. Because of the inherent gene expression similarity between the later cardiomyocyte differentiation time points, further investigation is warranted to determine whether the different key marker genes or the constructed gene expression patterns would better differentiate these time points in all datasets.
Differential gene expression analysis of high-throughput RNA-seq gene expression data represents a challenge, as datasets grow to several time points or conditions containing many biological replicates. We have developed a devGEA bioinformatics software suite that combines basic statistical analysis techniques, data visualization, and novel gene expression signature classification methods to improve the analysis of developmental time series. Additionally, we demonstrated the effective use of our methods in classifying developmental signatures that represent key stages of cardiomyocyte differentiation by filtering genes based on statistical descriptions and correlation to specific gene signatures. The robustness of the software and analysis was validated via RT-qPCR analysis of gene expression using the commercially available Gibco hiPSC cardiomyocyte differentiation procedure.
The devGEA analysis of directed hiPSC-cardiomyocyte differentiation provides an effective methodology for identifying and exploring gene expression signatures that characterize cardiac development, which is applicable to other domains of research. Three different classification methods used either basic statistics or correlations to identify similarly expressed genes. Additional experiments can be used to enhance or validate data analysis.
The GSE85331 and GSE122380 datasets offer a means to robustly identify gene expression signatures across cardiomyocyte differentiation and to explore the effectiveness of each method. To validate these findings, 32 genes comprising differentiation markers and understudied genes of the 3259 priority gene list were evaluated using RT-qPCR profiling of the cardiomyocyte differentiation of the GIBCO hiPSC line when using stringent criteria requiring the same gene signatures to be called in all three datasets across both experiments. Thirteen of the 32 genes were validated in this manner, indicating the reliability of signature identification between these diverse methods. Of the remaining genes on the 32 gene list, most were mid or late-cardiac genes, where at least one of the six classifications placed the gene in an adjacent cluster (i.e., classifying mid genes as late or late genes as mid). The discrepancies between these experiments may be due to the biological nature of cardiomyocyte differentiation or the technical challenges of discriminating unique developmental expression signatures characterizing a differentiating subpopulation from a heterogeneous population of cells. Nevertheless, our RT-qPCR investigation of important developmental cardiac markers from the correlation analysis demonstrated the robust classification of differentially expressed genes into specific cardiac developmental signatures.
Furthermore, these experiments identified unique gene expression patterns occurring during human cardiomyocyte differentiation, with several of these genes associated with mammalian heart development or congenital cardiovascular defects.
The earliest cell state transition involves specification of the mesodermal lineage from the pluripotency state. The mesodermal signature genes were transiently upregulated on day 2 and then repressed during the remainder of cardiomyocyte differentiation. The cadual-type homeobox genes CDX1 and CDX2, are early embryonic genes that control the anterior-posterior patterning of the developing embryo.43,44 Recent studies in mice have identified important roles for CDX genes in early embryonic fate decisions that drive differentiating cells to the cardiac lineage and in epicardial development.45,46 Additional genes associated with the mesodermal signature include msh homeobox MSX1 and doublesex and mab-3 related DMRT1 transcription factors. MSX1 and MSX2 double homozygous null mutant mice developed abnormalities in the atrioventricular valves and myocardium.47 Two additional genes include the CDK/ABL kinase interaction protein CABLES1 and the interleukin 17 receptor IL17RD which are yet to be investigated in heart development.
The early cardiomyocyte signature is characterized by transcriptional upregulation at day 2, with persistent or increasing gene expression persisting throughout the entire cardiomyocyte differentiation process. Examples of genes classified as early cardiomyocyte signatures include the Wnt cargo receptor WLS, YY1-associated factor YAF2, and transcriptional regulators ZEB2 and ZNF503. WLS encodes a conserved protein necessary for WNT secretion, which is required for early embryonic axis and heart field formation.48–50 While developmental functions for YAF2, ZEB2, and ZNF503 have been identified in mice, embryonic cardiac developmental roles for these genes are yet to be reported.51–53
Mid-cardiomyocyte genes represent the induction of gene expression on day four of cardiomyocyte differentiation. The protein kinase PKDCC gene and frizzled-class WNT receptor FZD4 are examples of candidate genes that have yet to be investigated for cardiac developmental functions. PKDCC is an essential mesenchymal regulator of lung development.54,55 FZD4 is expressed within the embryonic vasculature, and mutations are responsible for the incomplete vascularization of the retina occurring in human exudative vitreoretinopathy I disorder.56,57 Furthermore, FZD4 was recently found to be a marker of the lateral plate mesoderm and to contribute to cardiomyocytes and other cardiac cell lineages.58,59
Late-cardiomyocyte signature candidates are associated with later stages of differentiation involving myofibril organization and contractility. The actinin ACTN2, cardiomyopathy-associated protein CMYA5, sarcomeric protein CSRP3, nuclear lamin-interacting protein MLIP, sarcomeric protein MYOM1, and actin-binding protein NEXN were markedly enriched at the endpoint of the developmental series. Interestingly, each of these genes is associated with a human cardiomyopathy disorder, produces cardiomyopathy in a mouse model, or localizes to the cardiac myofibrils.60–64 Additionally, protein kinase HIPK3 and protein phosphatase regulatory subunit PPP1R12B were classified as late-cardiac signature genes. HIPK3 is expressed in the early developing heart; however, the function of the gene may be dispensable because homozygous null adult mice survive into adulthood and develop impaired insulin secretion and glucose tolerance.65,66 The PPP1R12B gene encodes a subunit of MYPT2, a muscle phosphatase that regulates cardiac myosin within contractile myofibrils.67
Overall, devGEA provides an effective analysis tool that enables the reproducible identification of unique cardiac signatures occurring throughout hiPSC-cardiomyocyte differentiation. Given the reproducibility of several genes across multiple experiments and cell lines, these genes may represent important regulatory nodes in the human developmental cardiac network, warranting further investigations. The study of heart development is of critical importance to biomedical, clinical, and translational research, as it allows us to understand and intervene in human cardiovascular disease. Directed cardiomyocyte differentiation from hiPSCs allows researchers to answer important biological questions regarding the differentiation of these specialized cells, the pathogenesis underlying the disease, and their potential therapeutic treatment for human cardiovascular disease. Since differential gene expression during hiPSC-directed cardiomyocyte differentiation recapitulates the in vivo cardiogenic program of the embryo, this experimental system allows us to advance and expand our knowledge of gene regulation in human cardiogenesis from other model organisms such as mice. The translational medicine effectiveness of this technique is already being realized by the generation and differentiation of hiPSCs into cardiomyocytes from patients with congenital cardiovascular disease, which allows the determination of the molecular, cellular, developmental, and physiological dysfunction underlying gene mutations.68,69 Additionally, hiPSCs can be genetically modified using RNAi, homologous recombination, or Crispr/Cas9 techniques to inactivate gene function during cardiomyocyte differentiation, allowing the study of gene function in the human developmental context. High-throughput gene expression profiling of hiPSC differentiation allows the classification of discrete gene expression patterns during cardiomyocyte differentiation, which may prove useful for the analysis of genetically modified hiPSCs and patient-derived hiPSCs that disrupt normal cardiac regulatory gene function.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)