Keywords
Cardiotoxicity, Doxorubicin, Ezetimibe, inflammation, oxidative stress
Cardiotoxicity, Doxorubicin, Ezetimibe, inflammation, oxidative stress
Cancer is a prevalent systemic disease characterized by uncontrolled cell proliferation and distant invasion, resulting in multiple organ damage, and ultimately, leading to a high rate of morbidity and mortality worldwide.1,2
One of the most important anticancer drugs in clinical practice is doxorubicin (DOX), also known as adriamycin, which belongs to the family of anthracycline antibiotics.3 Since its approval, DOX has been utilized in treating different types of cancer, including solid tumors, like breast, ovarian, testicular, thyroid, lung, liver, stomach and bladder cancers, as well as soft tissue sarcomas. Moreover, the drug has been employed in the treatment of hematologic malignancies, such as multiple myeloma, acute lymphoblastic leukemia, Hodgkin’s and non-Hodgkin’s lymphomas, in addition to certain childhood cancers, including neuroblastoma, osteosarcoma, Ewing’s sarcoma and rhabdomyosarcoma.1,4
Unfortunately, despite its broad antitumor spectrum, the clinical application of DOX is often limited by its irreversible and dose-dependent cardiotoxicity, which can result in left ventricular dysfunction (LVD) and heart failure (HF).5,6 On top of that, it has been estimated that the risk of LVD is approximately 7–26% when the cumulative dose of DOX reaches 550 mg/m2, and about 18–48% at a cumulative dose of 700 mg/m2.7
Cardiotoxicity that is induced by DOX seems to be a multifactorial and complex process, and there has been a long list of proposed mechanisms in this regard,8 one of which is the ability of DOX to generate excessive reactive oxygen species (ROS) and reactive nitrogen species (RNS) to the point that they would overpower the antioxidant defense system, causing disruption of cellular hemostasis and activation of the inflammatory response pathways, eventually leading to cell death,9,10 and from this point of view, developing a strategy that can hinder the development of the aforementioned events would be a possible approach to alleviate DOX-induced cardiotoxicity (DIC).
Ezetimibe (EZE) is a cholesterol-lowering drug that inhibits the absorption of dietary and biliary cholesterol from the small intestine by blocking the Niemann-Pick C1-Like protein (NPC1L1), which is a sterol transporter found on the brush border membrane of the intestinal epithelium.11
Apart from its beneficial impact on lipid profile, EZE has been shown to have pleiotropic properties, including antioxidant and anti-inflammatory effects that are independent of its ability to inhibit NPC1L1.11,12 Therefore, the current study was conducted to assess the hypothesis that EZE can provide protection against DIC in rats, which to our knowledge, has not been studied yet, and to elucidate the mechanisms underlying EZE’s possible cardioprotective effect.
The current study followed the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines,13 and was commenced after receiving approval from the Research Ethics Committee of the College of Medicine, University of Baghdad (approval date: December 31, 2023; approval number: 03-31).
A total of 24 adult male Wistar rats, weighing 200–250 g each, were included in the present study. The animals were acquired from and maintained in the animal house of the Iraqi Center for Cancer Research and Medical Genetics under conditions of controlled temperature and relative humidity, as well as alternating 12-hour intervals of light and darkness. In addition, rats were supplied with commercial pellets and had free access to ad libitum water throughout the experiment, and were acclimatized for one week ahead of the study.
All efforts were made to ameliorate suffering of the experimental rats, including proper ventilation of the animal house, cleaning the animal cages every other day, careful handling of the animals throughout the experiment, use of effective anesthetic dose and animal euthanasia was carried out far away from the alive ones.
Rats included in the present study were randomly allocated into four groups of six and treated as follows:
Group I (control): rats received corn oil via oral gavage for 14 consecutive days.
Group II (DOX): rats received corn oil via oral gavage for 14 consecutive days, and a single intraperitoneal (IP) injection of 15 mg/kg DOX hydrochloride (Pfizer, Australia) on the 12th day of the experiment.
Group III (10 mg/kg EZE + DOX): rats received 10 mg/kg EZE (Meryer, China) dissolved in corn oil via oral gavage for 14 consecutive days, and a single IP injection of 15 mg/kg DOX hydrochloride on the 12th day of the experiment.
Group IV (20 mg/kg EZE + DOX): rats received 20 mg/kg EZE dissolved in corn oil via oral gavage for 14 consecutive days, and a single IP injection of 15 mg/kg DOX hydrochloride on the 12th day of the experiment.
After 24 hours from final administration, the experimental rats were anesthetized using a combination of 150 mg/kg ketamine (Bremer Pharma GmbH, Germany) and 10 mg/kg lidocaine hydrochloride (GF Bonn GmbH, Germany). Then, for each rat, an abdominal incision was made and the inferior vena cava was exposed in order to collect a blood sample for biochemical analysis, after which the heart was excised, washed with cold phosphate-buffered saline (Sigma, USA) to get rid of any residual blood and bloated on filter paper. Following that, heart tissue samples were obtained for biochemical and gene expression analyses.
Each rat blood sample was immediately transferred into a serum separator tube (gel and clot activator tube), then the tubes were placed in a test tube rack and the blood was left to clot at room temperature for 30 min. Next, the tubes were centrifuged at 3500 rpm for 20 min, after which each sample supernatant layer, which represents the serum, was transferred into a pre-chilled microcentrifuge tube and preserved at −20 °C until utilization for the biochemical analysis.
For the biochemical analysis, the heart tissue samples were homogenized in an extraction medium that involved a combination of T-PER™ Tissue Protein Extraction Reagent (Thermo Fisher Scientific, USA) and Halt™ Protease Inhibitor Cocktail, EDTA-Free (Thermo Fisher Scientific, USA). Following their manufacturers’ instructions, enough working solution of the tissue protein extraction reagent and the protease inhibitor cocktail was prepared for all samples, then the tissue samples were weighed and placed in microcentrifuge tubes, after which the appropriate volume of the prepared working solution was added to each sample, with subsequent homogenization using a tissue grinding pestle and centrifugation at 10,000 rpm for 5 min to pellet tissue debris. Lastly, each sample supernatant layer was transferred into a pre-chilled fresh microcentrifuge tube and stored at −20 °C until use.
As for the gene expression analysis, the tissue samples were lysed by placing them in microcentrifuge tubes containing 1 mL TRIzol™ Reagent (Thermo Fisher Scientific, USA) and homogenized using tissue grinding pestles, after which the tubes were incubated for 5 min to allow complete dissociation of the nucleoprotein complex. Then, 0.2 mL chloroform was added to each tube, followed by incubation for 2–3 min and centrifugation for 10 min at 12,000 rpm. Subsequently, each tube mixture was separated into a lower red phenol-chloroform organic phase, an interphase, and a colorless upper aqueous phase that contains total RNA.
The next step was to transfer each tube aqueous phase into a new microcentrifuge tube. Then, 0.5 mL isopropanol was added to each tube in order to precipitate total RNA, after which the tubes were incubated for 10 min at 4°C and centrifuged for 10 min at 12,000 rpm, with subsequent precipitation of total RNA in the form of a white gel-like pellet at the bottom of the tubes.
Afterwards, for each tube, the supernatant layer was discarded, and 1 mL of 75% ethanol was added to resuspend the RNA pellet. Next, the tubes were vortexed briefly and centrifuged for 5 min at 7500 rpm, after which the supernatant layer of each tube was discarded, and the RNA pellet was air-dried for 10 min. Thereafter, 50 μL RNase-free water was added to each tube to solubilize the RNA pellet, followed by incubation in a heat block set at 55°C for 10 min. Finally, the tubes were stored at −20 °C until use.
Serum levels of cardiac injury biomarkers, including cardiac troponin I (cTnI) and N-terminal pro-B-type natriuretic peptide (NT-proBNP), as well as levels of oxidative stress biomarkers, including superoxide dismutase (SOD), glutathione peroxidase (GPx) and malonaldehyde (MDA), in addition to the inflammatory biomarker nuclear factor-kappa B (NF-κB) in the heart tissue homogenates were analyzed by means of enzyme-linked immunosorbent assay (ELISA) technique, using their commercially available kits (Cloud-Clone Corp, USA) and following the manufacturers’ instructions.
Two-step reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to analyze the relative mRNA expression of proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) in cardiac tissues, and all procedures were performed following the manufacturers’ protocols. At first, for each heart tissue sample, the isolated RNA was converted to cDNA with the aid of GoScript™ Reverse Transcription System (Promega, USA), after which the synthesized cDNA was quantified utilizing QuantiFluor® dsDNA System (Promega, USA) for downstream application. Moving forward, for each gene expression analysis, a mixture consisting of GoTaq® qPCR Master Mix (Promega, USA), cDNA as well as the gene forward and reverse primers (Macrogen, South Korea) was assembled to perform qPCR, provided that the housekeeping gene, beta-actin (β-actin), was employed to normalize mRNA expression levels of the target genes, and all primer sequences are listed in Table 1.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| TNF-α | ACCTTATCTACTCCCAGGTTCT | GGCTGACTTTCTCCTGGTATG |
| IL-1β | GACTTCACCATGGAACCCGT | GGAGACTGCCCATTCTCGAC |
| β-actin | AGGAGTACGATGAGTCCGGC | CGCAGCTCAGTAACAGTCCG |
Following that, qPCR was carried out utilizing a magnetic induction cycler (MIC) (Bio Molecular Systems, Australia), and the cycling conditions involved initial denaturation at 95 °C/5 min, followed by 40 cycles of denaturation at 95 °C/20 s, annealing at 55 °C/20 s, 65 °C/20 s and 60 °C/20 s for TNF-α, IL-1β and β-actin primers respectively, with subsequent extension at 75 °C/20 s.
After cycling had been completed, the amplification curves obtained for the three genes were analyzed utilizing micPCR software (version 2.10.0), in which thresholds and cycling threshold (Ct) values were automatically determined. Finally, the 2−ΔΔCt method was used to calculate the fold changes in mRNA expression of TNF-α and IL-1β relative to the control group.
Statistical analysis was performed utilizing GraphPad Prism software (version 9.5.1), in which the numeric data obtained from the study, presented as mean ± standard error of the mean (SEM), were analyzed by one-way analysis of variance (ANOVA) to evaluate the statistical significance of the results, followed by Tukey’s Post Hoc test for multiple comparisons, and P values of less than 0.05 were considered to be statistically significant.
As shown in Table 2 and Figure 1, the means of serum cTnI and NT-proBNP levels in DOX-treated rats were significantly elevated in compression with those of the control group. In contrast, treatment with either 10 or 20 mg/kg EZE prior to DOX administration significantly decreased the means of the earlier mentioned cardiac injury biomarkers compared with those of DOX group.14
| Group | cTnI (pg/mL) | NT-proBNP (pg/mL) |
|---|---|---|
| Control | 137.74 ± 12.00 | 0.67 ± 0.12 |
| DOX | 564.32 ± 58.71a**** | 4.91 ± 1.01a**** |
| 10 mg/kg EZE + DOX | 197.71 ± 15.54b**** | 0.91 ± 0.10b*** |
| 20 mg/kg EZE + DOX | 156.27 ± 16.62b**** | 0.77 ± 0.07b**** |
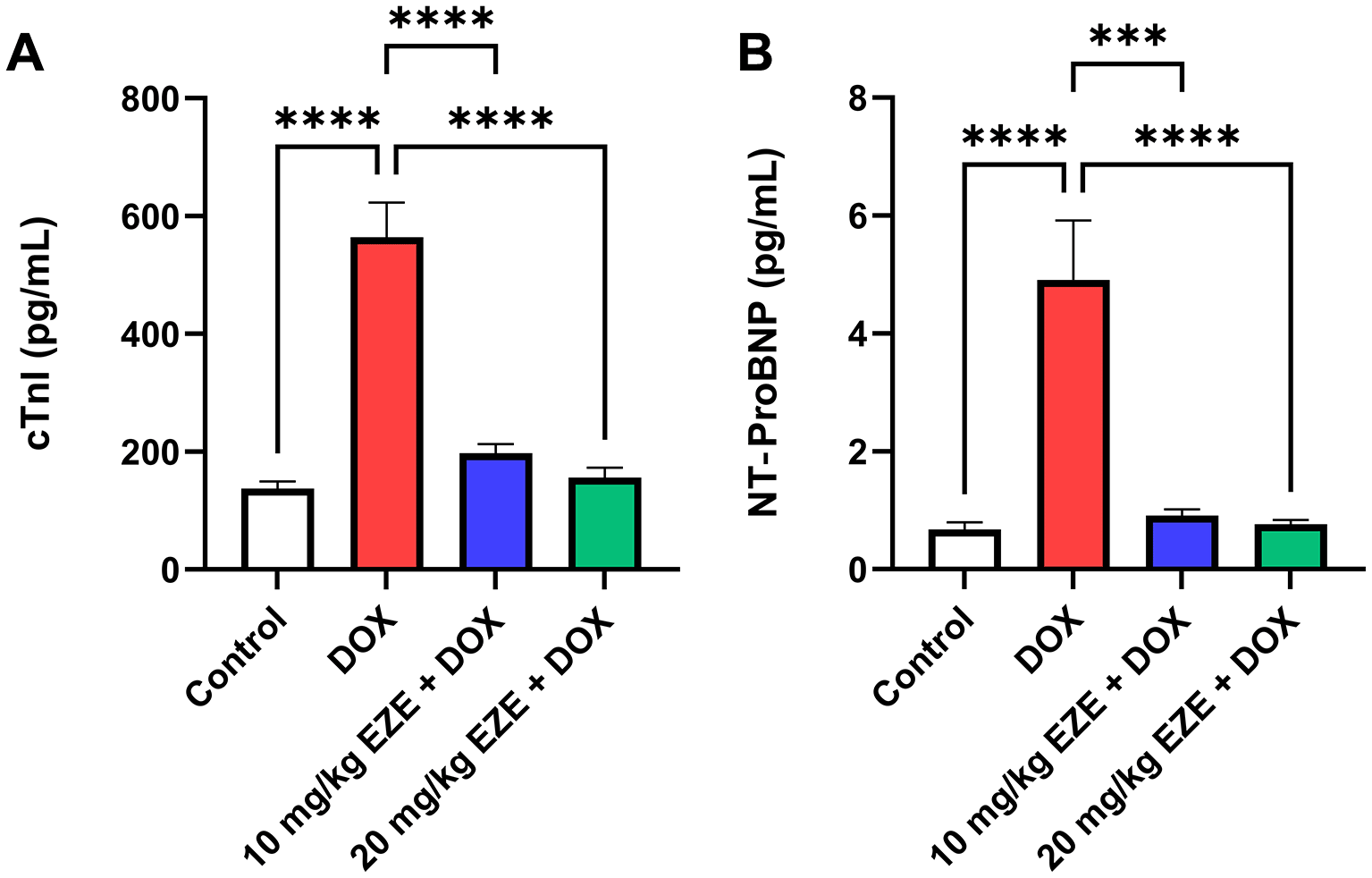
Serum levels of (A) cTnI and (B) NT-proBNP. Data are presented as mean ± standard error of the mean (SEM), n = 6. ***p < 0.001; ****p < 0.0001. cTnI, cardiac troponin I; DOX, doxorubicin; EZE, ezetimibe; NT-proBNP, N-terminal pro-B-type natriuretic peptide.
DOX administration resulted in a significant depletion in the means of cardiac SOD and GPx levels, as well as a significant increment in the mean of cardiac MDA levels compared with their corresponding values in the control group. On the other hand, the means of cardiac SOD and GPx levels were significantly higher, while the mean of cardiac MDA levels was significantly lower in rats that were treated with either 10 or 20 mg/kg EZE before receiving DOX than in those that received only DOX (Table 3 and Figure 2).14
| Group | SOD (ng/mL) | GPx (ng/mL) | MDA (ng/mL) |
|---|---|---|---|
| Control | 9.57 ± 0.85 | 103.06 ± 5.37b | 2.18 ± 0.55 |
| DOX | 2.23 ± 0.43a**** | 28.11 ± 6.55a**** | 12.36 ± 1.92a**** |
| 10 mg/kg EZE + DOX | 5.62 ± 0.60a*b* | 60.62 ± 2.64a**b* | 6.13 ± 0.74b** |
| 20 mg/kg EZE + DOX | 8.06 ± 1.22b*** | 92.50 ± 11.39b****c* | 3.46 ± 0.76b*** |
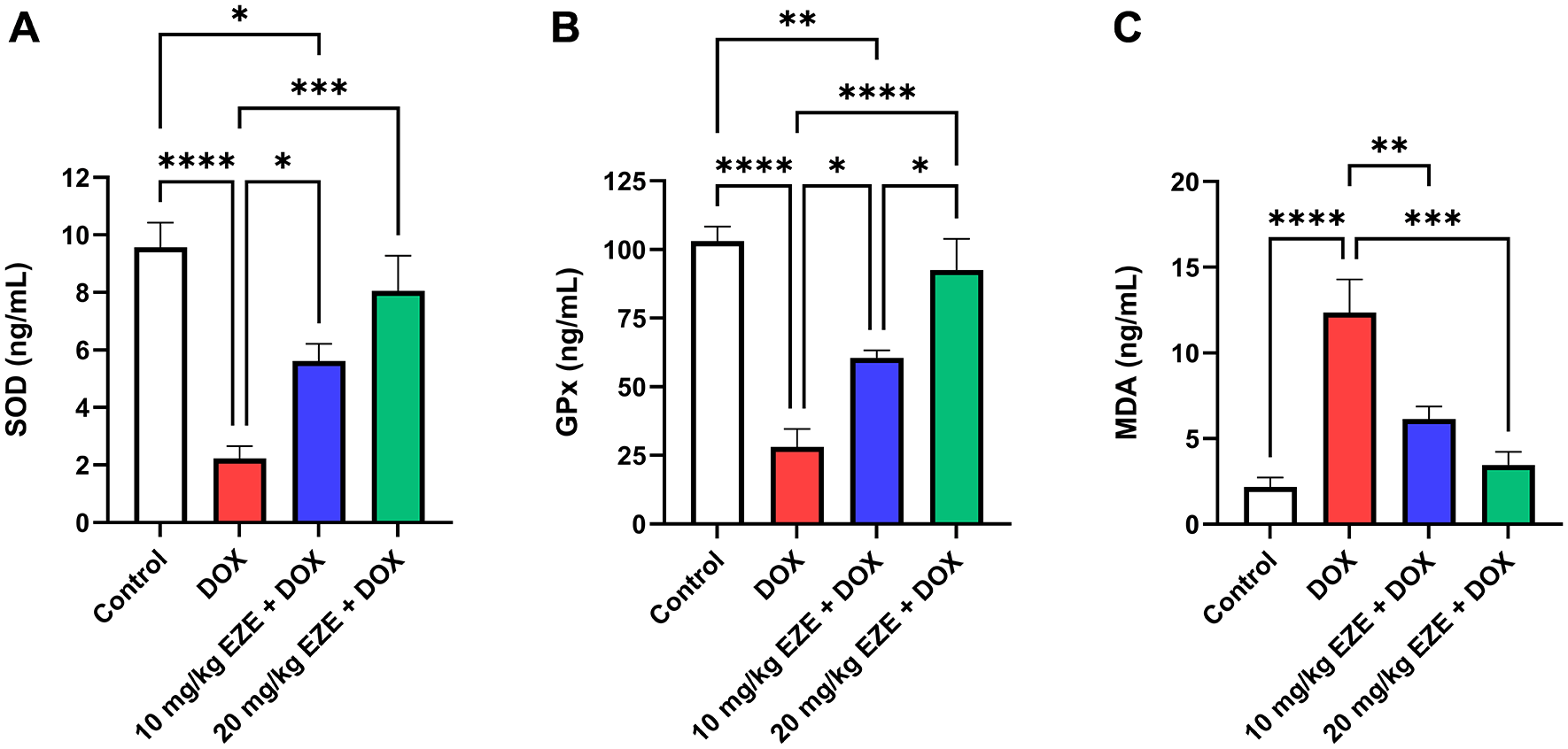
Levels of cardiac (A) SOD, (B) GPx and (C) MDA. Data are presented as mean ± standard error of the mean (SEM), n = 6. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. DOX, doxorubicin; EZE, ezetimibe; GPx, glutathione peroxidase; MDA, malonaldehyde; SOD, superoxide dismutase.
Treatment with DOX significantly elevated the mean of cardiac NF-κB levels when it was set up against that of the control group. On the contrary, rats that received either 10 or 20 mg/kg EZE ahead of DOX administration exhibited a significant decline in the mean of the earlier stated biomarker in comparison with that of DOX group, as presented in Table 4 and Figure 3.14
| Group | NF-κB (ng/mL) | TNF-α relative mRNA expression | IL-1β relative mRNA expression |
|---|---|---|---|
| Control | 4.45 ± 0.34 | 1.00 ± 0.47 | 1.00 ± 0.47 |
| DOX | 21.42 ± 4.74a*** | 3.38 ± 0.92a* | 6.18 ± 2.37a* |
| 10 mg/kg EZE + DOX | 5.99 ± 0.53b** | 0.93 ± 0.24b* | 7.26 ± 0.48a* |
| 20 mg/kg EZE + DOX | 5.26 ± 0.43b*** | 0.24 ± 0.09b** | 1.20 ± 0.51b*c* |

Levels of cardiac (A) NF-κB, (B) TNF-α relative mRNA expression and (C) IL-1β relative mRNA expression. Data are presented as mean ± standard error of the mean (SEM), n = 6. *p < 0.05; **p < 0.01; ***p < 0.001. DOX, doxorubicin; EZE, ezetimibe; IL-1β, interleukin-1 beta; NF-κB, nuclear factor-kappa B; TNF-α, tumor necrosis factor-alpha.
Additionally, a significant increment in the means of cardiac TNF-α and IL-1β relative mRNA expression levels was observed in DOX-treated rats compared with those of the control group. Meanwhile, pretreatment with either 10 or 20 mg/kg EZE caused a significant reduction in the mean of cardiac TNF-α relative mRNA expression levels compared with that of DOX-challenged rats. However, only pretreatment with 20 mg/kg EZE significantly diminished the mean of cardiac IL-1β relative mRNA expression levels in comparison with that of DOX group (Table 4 and Figure 3).14
One of the highly efficient antineoplastic agents with broad-spectrum application is the anthracycline antibiotic DOX. Nevertheless, the cardiotoxicity it causes represents a feared adverse effect and a potentially life-threatening response that can limit the clinical use of DOX and affect the quality of life of cancer patients, apart from their oncological prognosis.2,15
Results of the current study revealed that treating rats with a single IP injection of 15 mg/kg DOX hydrochloride caused a significant elevation in the means of serum levels of cardiac injury biomarkers, including cTnI and NT-proBNP, which is consistent with the results obtained from previous studies.16–18
Considering their high sensitivity, cardiac injury biomarkers are utilized for the early detection of cardiotoxicity, especially cardiac troponins, which are regulatory proteins that control calcium-mediated contractile function of the heart, and it has been demonstrated that newly elevated serum cTnI levels in patients receiving anthracyclines anticipate subsequent development of LVD.7,19,20 Additionally, NT-proBNP, which is secreted by cardiomyocytes in increased amounts during conditions of volume or pressure overload of the heart, is widely utilized in the detection of HF, and in terms of chemotherapy, its use can be helpful,7,21–23 based on studies that validated its role as a marker for the detection of DIC.24,25
The present study also showed that DIC was associated with notable changes in oxidative stress biomarkers in cardiac tissue, where the means of the antioxidant enzymes SOD and GPx were significantly depleted, while there was a significant increment in the mean of MDA contents, provided that MDA is a highly reactive dialdehyde derived from peroxidation of polyunsaturated fatty acids by means of reactive species.26,27
The abovementioned outcomes are compatible with those acquired from prior studies,17,28,29 which reinforces the theory that one of the major mechanisms underlying the development of DIC is oxidative stress, and the latter can be defined as a state of imbalance in which the production of ROS, such as superoxide anion (O2• –), hydrogen peroxide (H2O2) and hydroxyl radical (HO•), as well as RNS, such as peroxynitrite anion (ONOO−), exceeds the capacity of the endogenous antioxidant system, resulting in cellular damage.10,30,31
One of the reasons that makes the heart particularly susceptible to injury by DOX-induced oxidative stress is that antioxidant enzymes, including SOD, catalase (CAT) and GPx are less abundantly expressed in cardiomyocytes as compared with other cell types. Another reason is that DOX has a tendency to accumulate in the mitochondria, and since the heart is the body’s most metabolically active organ, it tends to have the highest mitochondrial content, rendering the cardiac cell the most vulnerable to oxidative damage.5,32
Studies elucidated that DOX induces oxidative stress as the quinone moiety in its structure undergoes one-electron reduction through the action of NADH- or NADPH-dependent enzymes to form a semiquinone radical, which tends to react rapidly with an oxygen molecule (O2), where it donates an electron to O2 to form O2• –, while recycling itself back to the quinone form, and this cycle is referred to as the redox cycling of DOX. Subsequently, the generated O2• – can be transformed by SOD into H2O2 that can be converted either into O2 and water (H2O) by means of CAT or GPx, or into a highly reactive and toxic HO• in the presence of ferrous ion (Fe2+). One more point to consider is that O2• – can also react with nitric oxide (NO) to form the RNS ONOO−. Eventually, overproduced HO• and ONOO− interact with the biomolecules nearby, causing lipid peroxidation, protein modification, DNA damage as well as disruption of mitochondrial function,33–35 as shown in Figure 4.
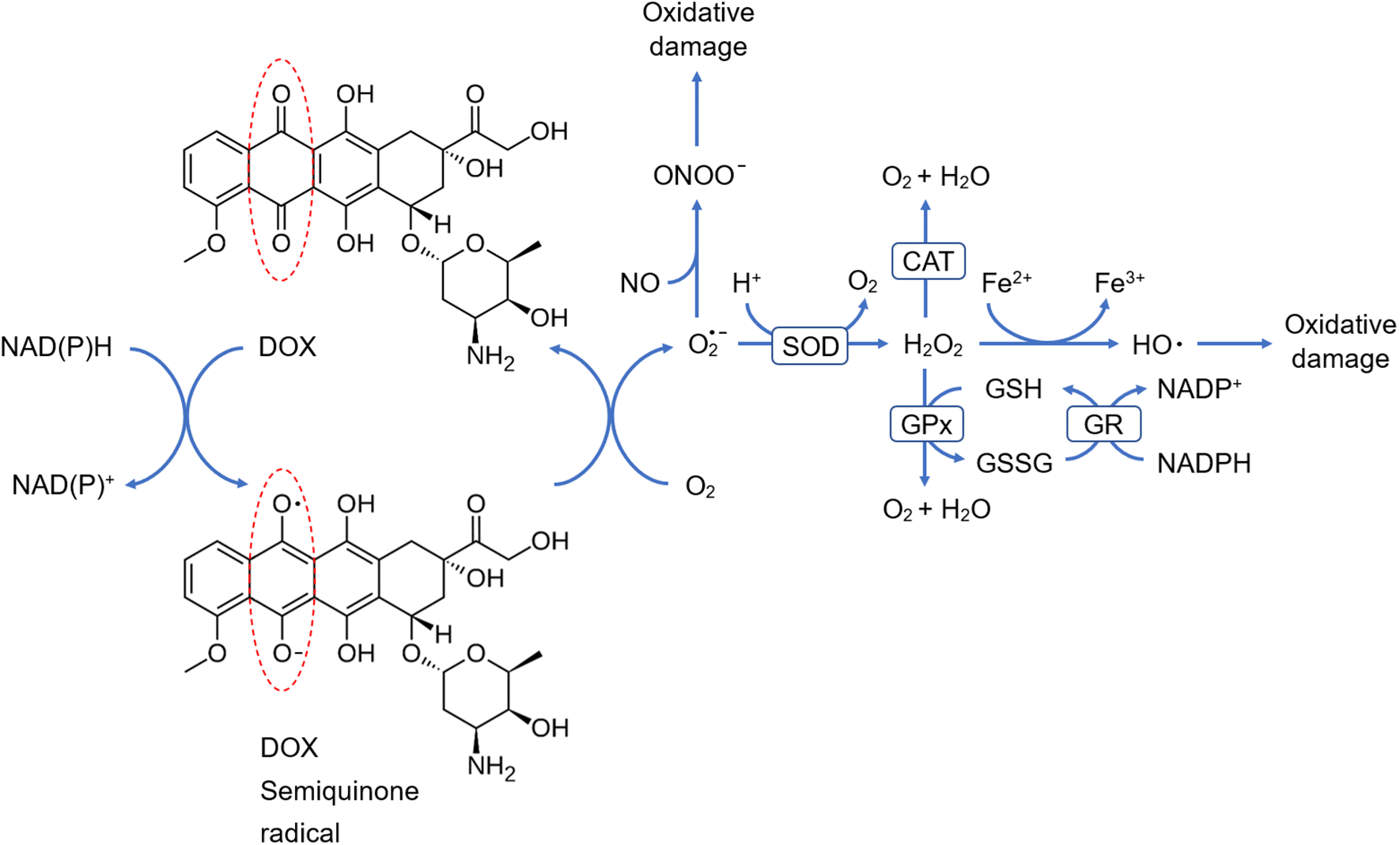
CAT, catalase; DOX, doxorubicin; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, glutathione disulfide; NAD+, nicotinamide adenine dinucleotide; NADP+, nicotinamide adenine dinucleotide phosphate; NADH, reduced nicotinamide adenine dinucleotide; NADPH, reduced nicotinamide adenine dinucleotide phosphate; SOD, superoxide dismutase. Chemical formulas: Fe+2, ferrous iron; Fe+3, ferric iron; H+, proton; H2O, water; H2O2, hydrogen peroxide; HO•, hydroxyl radical; NO, nitric oxide; O2, oxygen; O2• −, superoxide anion; ONOO −, peroxynitrite anion.
Our results also manifested that DOX administration significantly increased the means of cardiac NF-κB levels and its downstream proinflammatory cytokines, including TNF-α and IL-1β relative mRNA expression levels, and these observations are in agreement with those reported in other studies,36–38 thereby supporting another theory through which DOX induces cardiotoxicity, and that is inflammation.
Researchers pointed out that inflammation in DIC occurs as a result of NF-κB activation in response to oxidative stress,10,39 provided that NF-κB is a family of DNA binding proteins that function as homo or heterodimers to mediate the expression of numerous genes involved in cell proliferation, inflammation, immunity and apoptosis.40,41 In its resting state, NF-κB presents in the cytoplasm as a complex with a member of inhibitory proteins known as inhibitors of NF-κB (IκΒs) that mask nuclear localization signals to prevent NF-κB from being imported into the nucleus. However, in response to stimulation, an IκB kinase (IKK) phosphorylates IκB to promote its ubiquitination and proteasomal degradation, after which NF-κB becomes freed from IκB and translocates into the nucleus, where it upregulates the expression of proinflammatory cytokines, such as TNF-α, IL-1β, IL-6 and others, with consequent development of cardiac inflammation that can lead to cardiac remodeling and dysfunction.39,42–44
In our research, we aimed to investigate whether EZE can safeguard against DIC in rats. Interestingly, treating rats with either 10 or 20 mg/kg EZE before DOX administration significantly mitigated the cardiotoxic potential of the anticancer drug, and that was evidenced by a significant decline in the means of serum cTnI and NT-proBNP levels compared with those of DOX group, and to our knowledge, the current research is the first to estimate serum levels of the two earlier mentioned biomarkers in rats after receiving EZE for the prevention of a cardiac pathological condition.
Moreover, results of the present study demonstrated that pretreatment with EZE at a dose of 10 or 20 mg/kg caused a significant increment in the means of cardiac SOD and GPx levels, along with a significant reduction in the mean of cardiac MDA levels compared with those of DOX group, and these outcomes emphasize that the drug possesses antioxidant effect, as it was reported in an in vitro study, where EZE protected endothelial cells against oxidative stress induced by oxidized low-density lipoproteins, in which a reduction in ROS formation, as well as an enhancement in SOD and CAT activities were observed.45
Our study also showed that in comparison with DOX-challenged rats, pretreatment with either 10 or 20 mg/kg EZE significantly decreased the means of cardiac NF-κB levels and TNF-α relative mRNA expression levels, while the 20 mg/kg dose was also capable of significantly reducing the mean of cardiac IL-1β relative mRNA expression levels, and these findings can be attributed to the anti-inflammatory effect of the drug that was mentioned in previous studies, including one that utilized THP-1 human monocytic cell line treated with water-soluble cholesterol to induce inflammation, where the results manifested that EZE treatment dramatically decreased the proinflammatory cytokine TNF-α production by silencing NF-κB signaling.46
The antioxidant and anti-inflammatory actions of EZE were also evidenced in other studies, and in this regard, a recent one pointed out that EZE alleviated acetic acid-induced ulcerative colitis in rats by hampering oxidative stress and inflammation.11 Also, in a transient middle cerebral artery occlusion model in rats, EZE attenuated oxidative stress and prevented neuroinflammation, resulting in an improvement in neurological outcome.12
The rationale behind EZE attenuation of oxidative stress and inflammation can be related to its ability to activate nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor that modulates the expression of genes involved in cellular defense against oxidative stress insults, including the ones encoding SOD, CAT, GPx and other antioxidants.47,48 Under basal conditions, Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1), a negative regulator of Nrf2 that mediates its ubiquitination and proteasomal degradation to prevent unwarranted expression of Nrf2-target genes. However, under stressed conditions, Nrf2 dissociates from Keap1 and translocates into the nucleus, where it upregulates the expression of antioxidant genes through binding to their antioxidant response element (ARE), which is an enhancer sequence placed at the promoter region of several cytoprotective genes. Consequently, the activity of antioxidants would be boosted, leading to suppression of oxidative stress-mediated activation of the NF-κB signaling pathway,49 as shown in Figure 5.
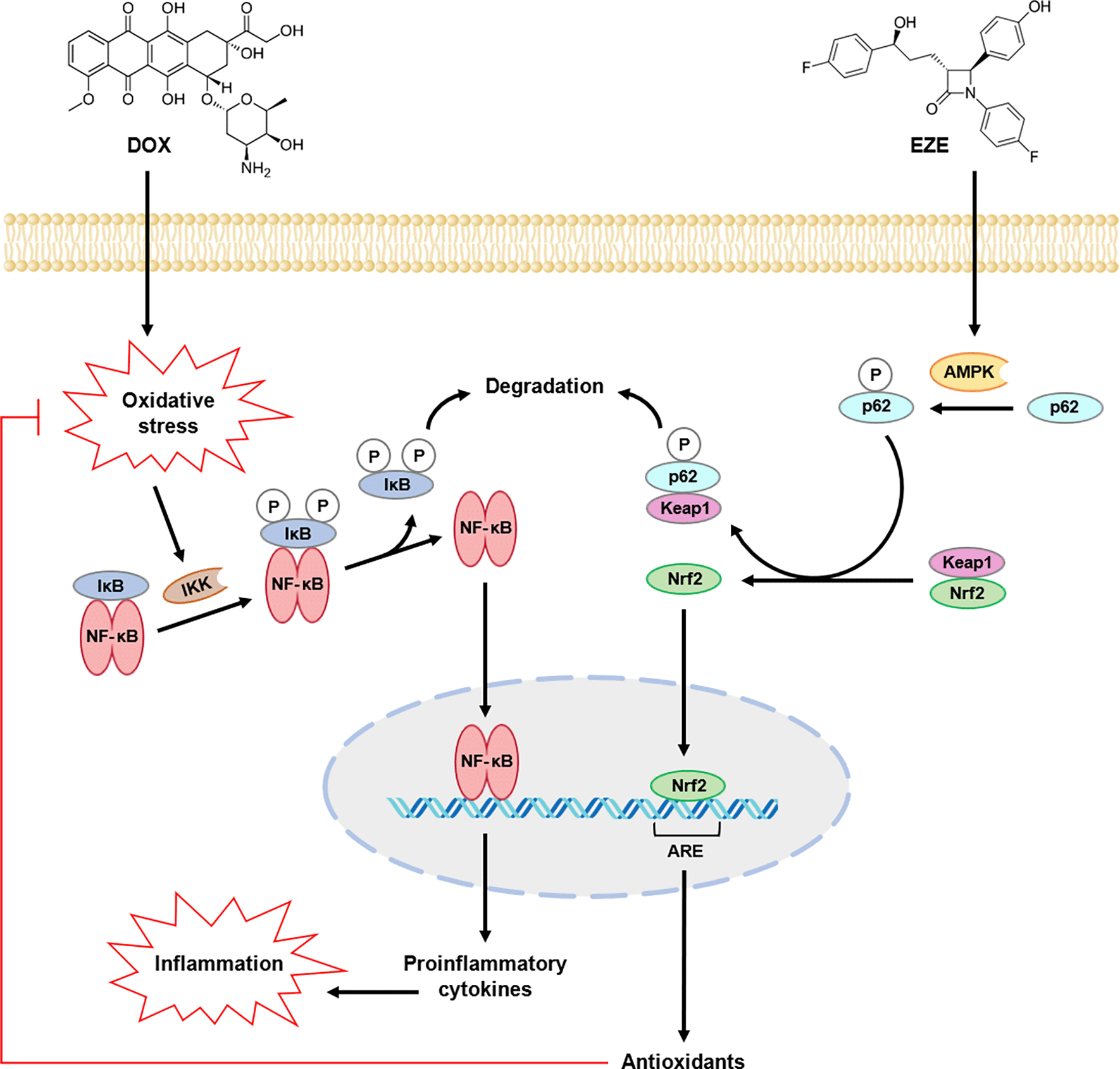
AMPK, adenosine monophosphate-activated protein kinase; ARE, antioxidant response element; DOX, doxorubicin; EZE, ezetimibe; IκB, inhibitor of nuclear factor-kappa B; IKK, IκB kinase; Keap1, Kelch-like ECH-associated protein 1; NF-κB, nuclear factor-kappa B; Nrf2, nuclear factor erythroid 2-related factor 2; P, phosphate.
The possible mechanism through which EZE induces Nrf2 activation has been elucidated in one study, where the drug was found to disrupt the interaction between Nrf2 and Keap1 through adenosine monophosphate-activated protein kinase (AMPK)-mediated phosphorylation of p62, an adaptor protein that functions as a central scaffold in various cellular processes, including autophagy. Accordingly, upon its phosphorylation, p62 binds to Keap1, resulting in autophagic degradation of the latter, after which the liberated Nrf2 translocates into the nucleus to upregulate the expression of antioxidant genes,48,50 and this can explain, at least in part, why EZE exerted a protective effect against DIC in rats (Figure 5).
The current research demonstrates, for the first time, that pretreatment with the cholesterol-lowering drug EZE can attenuate DIC in rats, as evidenced by a notable reduction in the serum levels of cardiac injury biomarkers. Also, we believe that the cardioprotective effect of EZE was mediated by suppression of DOX-induced oxidative stress and NF-κB-mediated inflammation, where EZE raised the levels of antioxidant enzymes, diminished lipid peroxidation and downregulated the gene expression of proinflammatory cytokines in cardiac tissues, thereby, it has the potential to serve as a promising candidate for alleviating DOX-induced cardiac injury.
The current study followed the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines,13 and was commenced after receiving approval from the Research Ethics Committee of the College of Medicine, University of Baghdad (approval date: December 31, 2023; approval number: 03-31).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)