Keywords
Acinetobacter baumannii, Klebsiella pneumoniae, MDR bacteria, Prophage annotation, Prophage detection
This article is included in the Antimicrobial Resistance collection.
Acinetobacter baumannii, Klebsiella pneumoniae, MDR bacteria, Prophage annotation, Prophage detection
Antibiotic resistance is a major public health concern worldwide (World Health Organization, 2023). Multidrug-resistant (MDR) bacterial infections are challenging to treat as these bacteria have developed resistance to a variety of conventional drugs. Overuse and misuse of antibiotics in both humans and livestock is a major factor that has contributed to the creation and spread of antibiotic-resistant bacteria (World Health Organization, 2023; Mittal et al., 2020). MDR infections in humans result in a wide range of diseases, including skin infections, pneumonia, sepsis, and urinary tract infections. These infections can be difficult to treat, leading to longer hospital stays, higher healthcare costs, and a higher mortality risk. The number of MDR infections is increasing globally, and in some severe cases, there are no viable or effective treatments available (Chinemerem et al., 2022).
Phage therapy utilizes viruses that selectively target and infect bacterial cells. This highly specific approach offers an effective alternative to antibiotics for the treatment of bacterial infections, without causing harm to eukaryotic cells. People in Eastern Europe and the former Soviet Union have used phage therapy to treat bacterial infections, with treatments initiated nearly a century ago (Chanishvili, 2012). In recent years, phage therapy has received interest as a potential solution to the problem of antibiotic resistance and phage cocktails with selective bacteriophages provide a promising strategy for treating infections caused by MDR bacterial strains (Yen et al., 2017; Michodigni et al., 2022).
The carbapenem-resistant strains of Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa are among the globally prevalent MDR bacteria that have caused bacterial outbreaks (Buehrle et al., 2017; Hamidian & Hall, 2011). Carbapenem-resistant Pseudomonas aeruginosa can cause pneumonia and bacteremia in infected subjects (Wen et al., 2018). Based on their clinical significance, these bacteria have been selected as targets for phage cocktail-based bacteriophage therapy (Adams et al., 2008; Cienfuegos-Gallet et al., 2022; Aly et al., 2008).
Prophages are the genomes of bacteriophages that are integrated into the chromosomes of host bacteria. Prophages remain dormant until they express their genomic regions in order to initiate a lytic cycle. They serve as a reservoir of genes that are required for the expression of structural and functional viral components that produce an active bacteriophage (Fortier and Sekulovic, 2013; Premetis et al., 2023). This study considered a phage cocktail creation that could tackle certain MDR bacteria. Instead of considering individual phages, a phage cocktail design was undertaken that contained a mixture of selected phages since this represents a potentially viable method for targeting a broader range of MDR bacterial strains.
Three whole genome sequences of bacteria were selected based on their pathogenicity in humans and generation of MDR diseases. The NCBI GenBank was used to obtain the nucleotide sequences of Acinetobacter baumannii AB0057 (accession number: NC_011586.1), Klebsiella pneumoniae subsp. pneumoniae HS11286 (accession number: NC_016845.1), and Pseudomonas aeruginosa UCBPP-PA14 (accession number: NC_008463.1).
The presence of prophages in the genomes of Acinetobacter baumannii AB0057, Klebsiella pneumoniae subsp. pneumoniae HS11286, and Pseudomonas aeruginosa UCBPP-PA14 were investigated by utilizing the results of prophage prediction from a previous study conducted by Mageeney et al. (2020a). In this study, prophage prediction was performed using two bioinformatics software programs: Islander (Hudson et al., 2015) and TIGER (Mageeney et al., 2020b). The predicted prophages were extracted from the output files generated by these tools and were used for further analyses. In addition, the PHASTER tool (Arndt et al., 2016) was also used to identify potential prophages in the selected bacterial genomes.
Open reading frames encoded by the 11 selected prophage sequences were predicted by two command line tools Prodigal Hyatt et al., 2010) and Phanotate McNair et al., 2019), and an online tool GeneMark.hmm (Besemer et al., 2001).
The Prokka annotation tool (Seemann, 2014) was utilized to locate the transfer RNAs, CDS, and ribosomal RNAs from the predicted prophages. In order to determine the locations of genomic features inside contigs, Prokka uses external feature prediction algorithms.
Predicted ORFs were annotated using the BLASTP algorithm against the non-redundant (nr) protein database of the NCBI (National Center for Biotechnology Information).
The putative promoter sequences and regions on the phage genomes were identified using BPROM which is a promoter online analysis tool of Softberry (Salamov and Solovyev, 2011).
Virulent genes on the predicted prophage sequences were detected by using the VFDB: Virulence Factor Database.
Rho-independent transcription terminators on predicted prophage sequences were detected using ARNold program (Naville et al., 2011).
tRNAs carried by prophage sequences were detected using the online program tRNAscan-SE (Schattner et al., 2005) and ARAGORN. The rRNA genes were also searched using Barrnap ribosomal RNA predictor.
A dot plot was generated using Gepard software in order to check the similarity between sequences of prophages (Krumsiek et al., 2007).
To identify the phages with the highest similarity, Blastn analysis was performed against a standard database of nucleotides within NCBI.
Comparative genome analysis of predicted prophages was conducted using EasyFig 2.2 (Sullivan et al., 2011). Genome maps of selected prophage sequences were constructed with their highest similar lytic phages using the tBlastx analysis and with a minimum selected e-value of 0.001.
Phylogenetic analysis of the selected phages was performed based on the nucleotide sequences of their large terminase subunits. A neighbor-joining phylogenetic tree was constructed for nucleotide sequences of finally selected prophages using MEGA 11 software with 1000 bootstrap replications (Tamura et al., 2021).
The detection of the presence and then annotation of prophages within bacterial genomes was undertaken. Similar to the study of Mageeney et al. (2020b), 7 prophages from Klebsiella pneumoniae subsp. pneumoniae HS11286, 3 prophages from Acinetobacter baumannii AB0057, and 1 prophage from Pseudomonas aeruginosa UCBPP-PA14 were detected using islander and TIGER algorithms. The island length and coordinates of these predicted prophages were determined and have been included in Table 1. Subsequently, whole genome sequences of Acinetobacter baumannii AB0057, Pseudomonas aeruginosa UCBPP-PA14, and Klebsiella pneumoniae subsp. pneumoniae HS11286 were retrieved from NCBI (accession numbers “NC_011586.1”, “NC_008463.1” and “NC_016845.1”, respectively) and prophage prediction undertaken using the PHASTER tool (Table 2).
About 29 ORFs were predicted from Acinetobacter baumannii AB0057 prophages (1464206-1483241), 79 ORFs from Acinetobacter baumannii AB0057 prophages (2759376-2809756), and 85 ORFs from Acinetobacter baumannii AB0057 prophages (2759376-2809756). Only 14 ORFs were predicted from Pseudomonas aeruginosa UCBPP-PA14 prophage (4345129-4355794). There were 14, 13, 73, 37, 63, 43, and 22 ORFs predicted from 7 Klebsiella pneumoniae subsp. pneumoniae HS11286 prophages (Table 3) and their positions, strand type and length of gene were also annotated (Underlying data: ORFs) (Nawaz, 2024).
The BLASTP algorithm was used to annotate ORFs in the prophage sequences of Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa. The sequences were compared with known protein sequences in the NCBI non-redundant protein database, and where possible, putative functions and biological roles were assigned (Underlying data: BLASTp_analysis) (Nawaz, 2024). A total of 29, 79, and 85 ORFs were identified in 3 prophages of Acinetobacter baumannii with maximum protein sequence identity percentages of 100% and minimum identity percentages ranging from 30.84 to 42.82%. Similarly, 14, 13, 73, 37, 63, 43, and 22 ORFs were identified in 7 prophages of Klebsiella pneumoniae with maximum protein sequence identity percentages of 100% and minimum identity percentages ranging from 27.51 to 93.83%. Finally, 14 ORFs were identified in 1 prophage of Pseudomonas aeruginosa, with maximum protein sequence identity percentages of 100% and minimum identity percentages of 38.22%. ORFs with their maximum and minimum identities were also predicted (Table 4).
Annotations of prophages revealed the coding sequences (CDS) and tRNAs carried by the prophage sequences. One tRNA was found from Acinetobacter baumannii AB0057 prophage (3311844-3364667) and two tRNAs from Klebsiella pneumoniae subsp. pneumoniae HS11286 prophage (1288317-1338719). However, no ribosomal RNAs were found in the prophage sequences (Table 5).
The three prophages of Acinetobacter baumannii had 47, 126, and 133 putative promoters, respectively. Among the seven prophages of Klebsiella pneumoniae, the number of putative promoters were 25, 19, 108, 67, 95, 78 and 37. For the single prophage of Pseudomonas aeruginosa, 19 putative promoters were identified (Table 3). The list of promoter sequences of 11 prophages with their transcription factors (TFs), binding site, position and binding scores were recorded (Underlying data: Promoter_sequences) (Nawaz, 2024).
The expression of virulent genes produces bacterial virulence factors, which have an essential role in bacterial pathogenicity (Abdulateef et al., 2023), hence, it is important to check for their existence in prophage sequences prior to prophage final selection. The prediction analysis identified peptidoglycan DD-metalloendopeptidase family protein and undecaprenyl-phosphate glucose phosphotransferase in one prophage sequence from Klebsiella pneumoniae, which are known virulence factors (Paczosa & Mecsas, 2016). Additionally, the prophage of Pseudomonas aeruginosa was found to contain phenazine biosynthesis protein PhzF, which is involved in phenazine biosynthesis and has been linked to virulence in P. aeruginosa (Blankenfeldt & Parsons, 2014) (Table 6).
| Bacterial strain | Detected prophage position | Size | tRNAs detected using ARAGORN and tRNAscan-SE | rRNA detected using Prokka and Barrnap | Virulence factors detected using VFDB | Terminator regions detected using Softberry BPROM | Promoter regions detected using ARNold | Dot plot alignment results from Gepard 2.1 |
|---|---|---|---|---|---|---|---|---|
| (nt) | (kb) | (n) | (n) | (n) | (n) | |||
| Acinetobacter baumannii AB0057 | 1464206-1483241 | 19.04† | 0 | 0 | N/A | 18 | 47 | - |
| 2759376-2809756 | 50.38 | 0 | 0 | N/A | 53 | 126 | N/A | |
| 3311844-3364667 | 52.82 | 1 | 0 | N/A | 61 | 133 | N/A | |
| Klebsiella pneumoniae subsp. pneumoniae HS11286 | 581777-594029 | 12.25† | 0 | 0 | Peptidoglycan DD-metalloendopeptidase family protein, undecaprenyl-phosphate glucose phosphotransferase‡ | 10 | 25 | - |
| 1062574-1073328 | 10.76† | 0 | 0 | N/A | 8 | 19 | - | |
| 1288317-1338719 | 50.40 | 2 | 0 | N/A | 35 | 108 | N/A | |
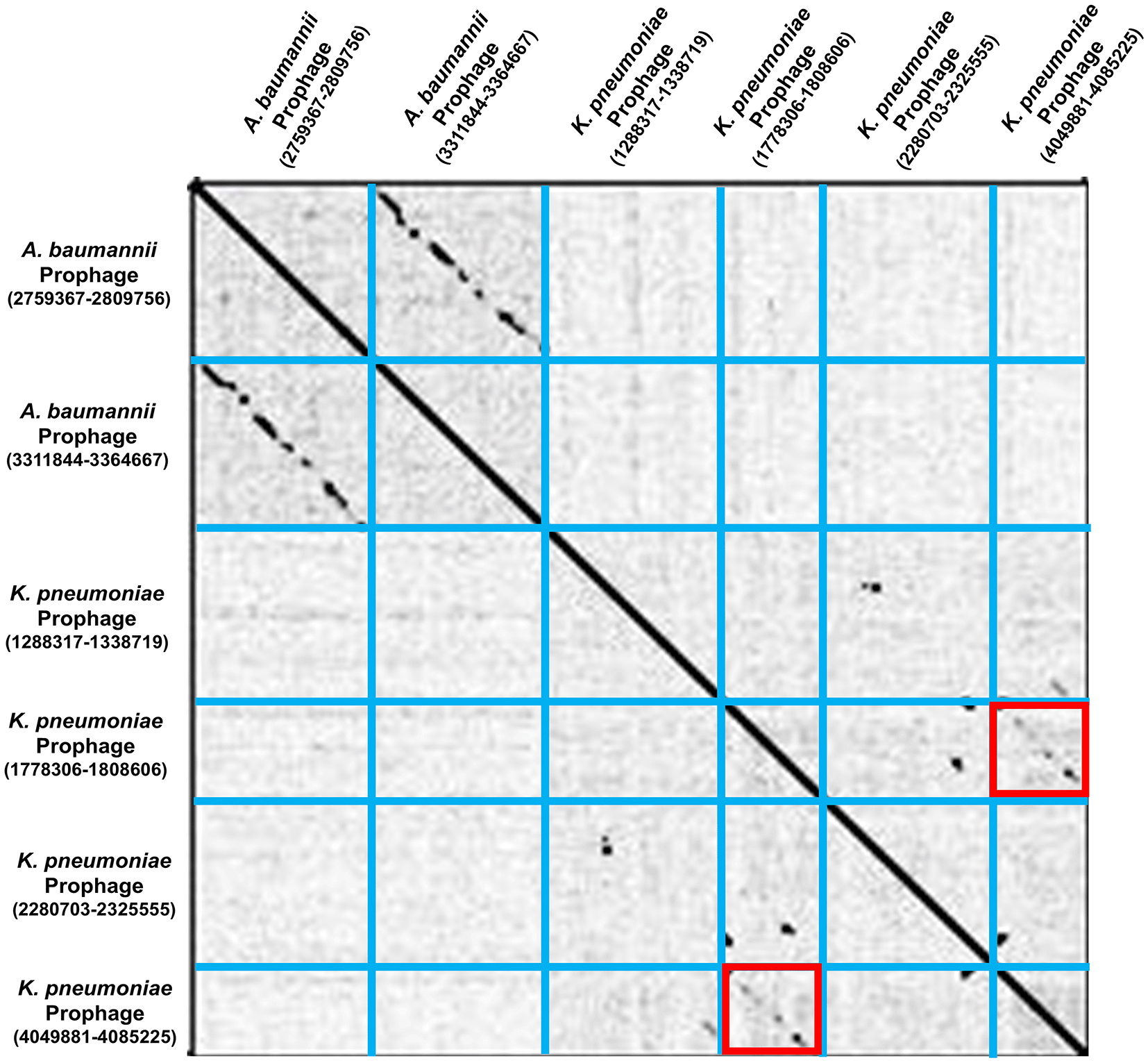
| 1778306-1808606 | 30.30 | 0 | 0 | N/A | 25 | 67 | Similarity found with (4049881-4085225)‡ | |
| 2280703-2325555 | 44.85 | 0 | 0 | N/A | 31 | 95 | N/A | |
| 4049881-4085225 | 35.34 | 0 | 0 | N/A | 29 | 78 | Similarity found with (1778306-1808606)‡ | |
| 4819189-4835185 | 16.00 | 0 | 0 | N/A | 9 | 37 | N/A | |
| Pseudomonas aeruginosa UCBPP-PA14 | 4345129-4355794 | 10.67† | 0 | 0 | Phenazine biosynthesis protein PhzF, isomerase‡ | 2 | 19 | - |
Rho-independent transcriptional termination in bacteria is characterized by the formation of a G/C rich hairpin structure which efficiently terminates transcription elongation without the requirement for a Rho-factor. The prediction of Rho-independent terminators is required to understand the transcriptional regulation of prophage genes and their integration into the host genome (Naville et al., 2011). Sequence analysis showed the presence of various Rho-independent transcription terminators in prophages, including 3 prophages of Acinetobacter baumannii containing 18, 53, and 61 terminators, 7 prophages of Klebsiella pneumoniae containing 10, 8, 35, 25, 31, 29, and 9 terminators, and 1 prophage of Pseudomonas aeruginosa containing 2 terminators (Table 3). The predicted transcription terminators, the position of terminators in the prophage genome, strand type, and the sequence of predicted terminators were also recorded (Underlying data: Terminators_sequences) (Nawaz, 2024).
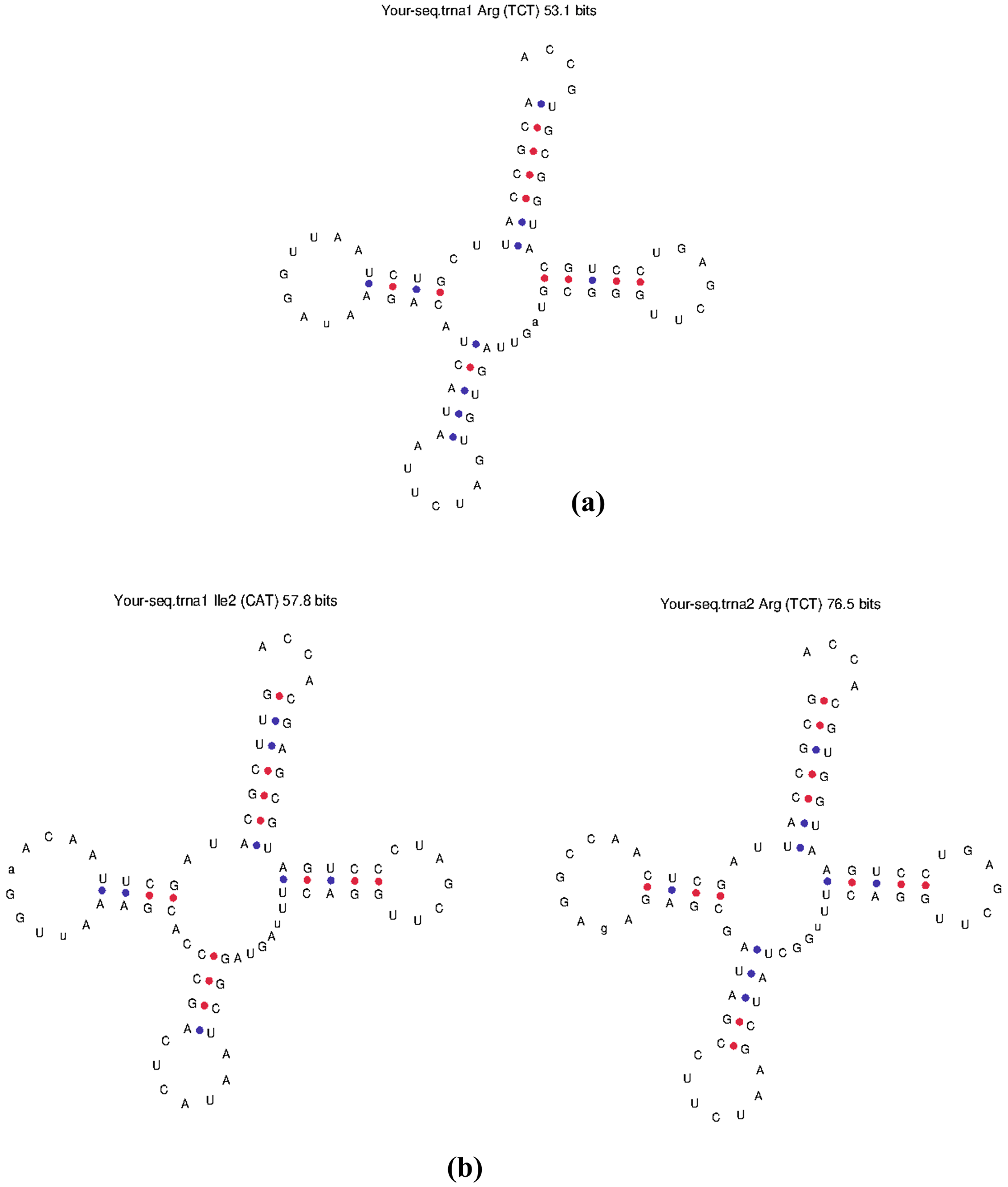
tRNAs prediction was undertaken using tRNA-SCAN-SE and ARAGORN programs and these detected the presence of 3 tRNA regions in the 11 prophage genomes (Table 5). Given that the host bacteria normally supply the tRNA molecules required for translation, this result raises the possibility that these prophages rely on the host translation machinery for protein synthesis. The structure of the tRNAs was also predicted (Figure 1a, b). Barrnap was used to attempt the detection of rRNA genes from the prophage sequences, but none were detected.
A dot plot, created using Gepard software, provided a means to investigate the similarity between sequences of the selected Acinetobacter baumannii AB0057 and Klebsiella pneumoniae subsp. pneumoniae HS11286 prophages (Figure 2). In order to avoid redundancy, the Klebsiella pneumoniae subsp. pneumoniae HS11286 prophage (1778306-1808606) was rejected from further consideration, as its sequence was found to be highly similar to that of Klebsiella pneumoniae subsp. pneumoniae HS11286 prophage (4049881-4085225).
For the selection of viable phages, the threshold values for annotations including prophage size, presence of any tRNAs, rRNAs, virulence factors, terminators and promoter regions, and sequence similarity analysis were taken into consideration. The prophages having sequence length less than 30 kb, those sharing sequence similarity with other prophages, or having any potential virulence factors were excluded from further study. Finally, based on the above-mentioned criteria, two out of three prophages from Acinetobacter baumannii AB0057 and three out of seven prophages from Klebsiella pneumoniae subsp. pneumoniae HS11286 were selected as viable phages (Table 6).
Comparative genome analysis of the 5 prophages with their highest similar lytic phage sequences were analysed using EasyFig 2.2. A multiple sequence alignment between our predicted prophage sequences and similar prophages was performed. A visual representation was produced showing linear comparisons for the similarities and differences between the prophages and their closest homologs. The genome map of Acinetobacter baumannii prophage (2759376-2809756) and Acinetobacter phage YC#06 (ON391949.1) indicated a similarity of 95.67% (Figure 3a), while A. baumannii prophage (3311844-3364667) with Acinetobacter phage Acba_3 (OQ101248) showed a similarity of 99.86% (Figure 3b). A higher similarity of 100% was indicated in the genome maps constructed for Klebsiella pneumoniae prophage (1288317-1338719) with Phage LAU1 (MN688132.1) (Figure 4a) and Klebsiella pneumoniae prophage (1778306-1808606) with Klebsiella phage ST437-OXA245phi4.2 (NC_049449.1) (Figure 4b). The genome map of Klebsiella pneumoniae prophage (2280703-2325555) with the highest similar Klebsiella phage ST11-VIM1phi8.3 (MK416019.1) showed a similarity of 99.99% (Figure 4c).


BLASTn analysis was performed using 5 selected prophages. The most identical phages were selected for further analysis to check the viability and lytic potential of the prophages. The sequence similarity between two prophages of Acinetobacter baumannii and previously known phage sequences ranged from 93 to 99%, and for the three prophages of Klebsiella pneumoniae, it ranged from 80 to 100%. The identical phages, sharing 99.9% and 100% similarity with our prophages, were found to have been previously used in phage therapy (Jeon et al., 2012; Bagińska et al., 2023; Lavigne et al., 2004). The sequences of identical phages were retrieved via their accession numbers (Table 7).
| Prophages | Similar phages | Accession numbers | Similarity (%) | Query coverage (%) |
|---|---|---|---|---|
| Acinetobacter baumannii (2759376-2809756) | Acinetobacter phage YMC/09/02/B1251_ABA_BP | NC_019541.1 | 96.06 | 40 |
| Acinetobacter phage Acba_1 | OQ101250.1 | 99.46 | 53 | |
| Acinetobacter phage Acba_11 | OQ101254.1 | 93.70 | 37 | |
| Acinetobacter phage Acba_15 | OQ101257.1 | 99.43 | 44 | |
| Acinetobacter baumannii (3311844-3364667) | Alcaligenes phage Piluca | MZ326864.1 | 94.30 | 4 |
| Acinetobacter phage Ab105-2phideltaCI404ad | MZ514874.1 | 87.18 | 47 | |
| Acinetobacter phage vB_AbaS_TRS1 | NC_031098.1 | 75.38 | 6 | |
| Acinetobacter phage Acba_3 | OQ101248.1 | 99.86 | 19 | |
| Klebsiella pneumoniae (1288317-1338719) | Klebsiella phage ST11-VIM1phi8.1 | MK448233.1 | 99.86 | 20 |
| Klebsiella phage ST899-OXA48phi17.1 | MK448236.1 | 96.07 | 43 | |
| Enterobacter phage LAU1 | MN688132.1 | 100 | 3 | |
| Klebsiella phage GZ7 | OP114732.1 | 98.62 | 12 | |
| Klebsiella pneumoniae (1778306-1808606) | Klebsiella phage ST11-VIM1phi8.3† | MK416019.1 | 99.99 | 88 |
| Klebsiella phage ST258-KPC3phi16.2† | MK433582.1 | 99.99 | 89 | |
| Klebsiella phage ST437-OXA245phi4.2† | NC_049449.1 | 100 | 89 | |
| Klebsiella phage ST512-KPC3phi13.2† | NC_049452.1 | 99.89 | 89 | |
| Klebsiella pneumoniae (2280703-2325555) | Klebsiella phage KPP5665-2 | MF695815.1 | 95.72 | 18 |
| Klebsiella phage ST11-VIM1phi8.3† | MK416019.1 | 99.99 | 78 | |
| Klebsiella phage Mulock | MN098327.1 | 97.25 | 34 | |
| Klebsiella phage ST512-KPC3phi13.5 | MN166823.1 | 99.98 | 56 | |
| Klebsiella phage GZ7 | OP114732.1 | 94.02 | 31 |
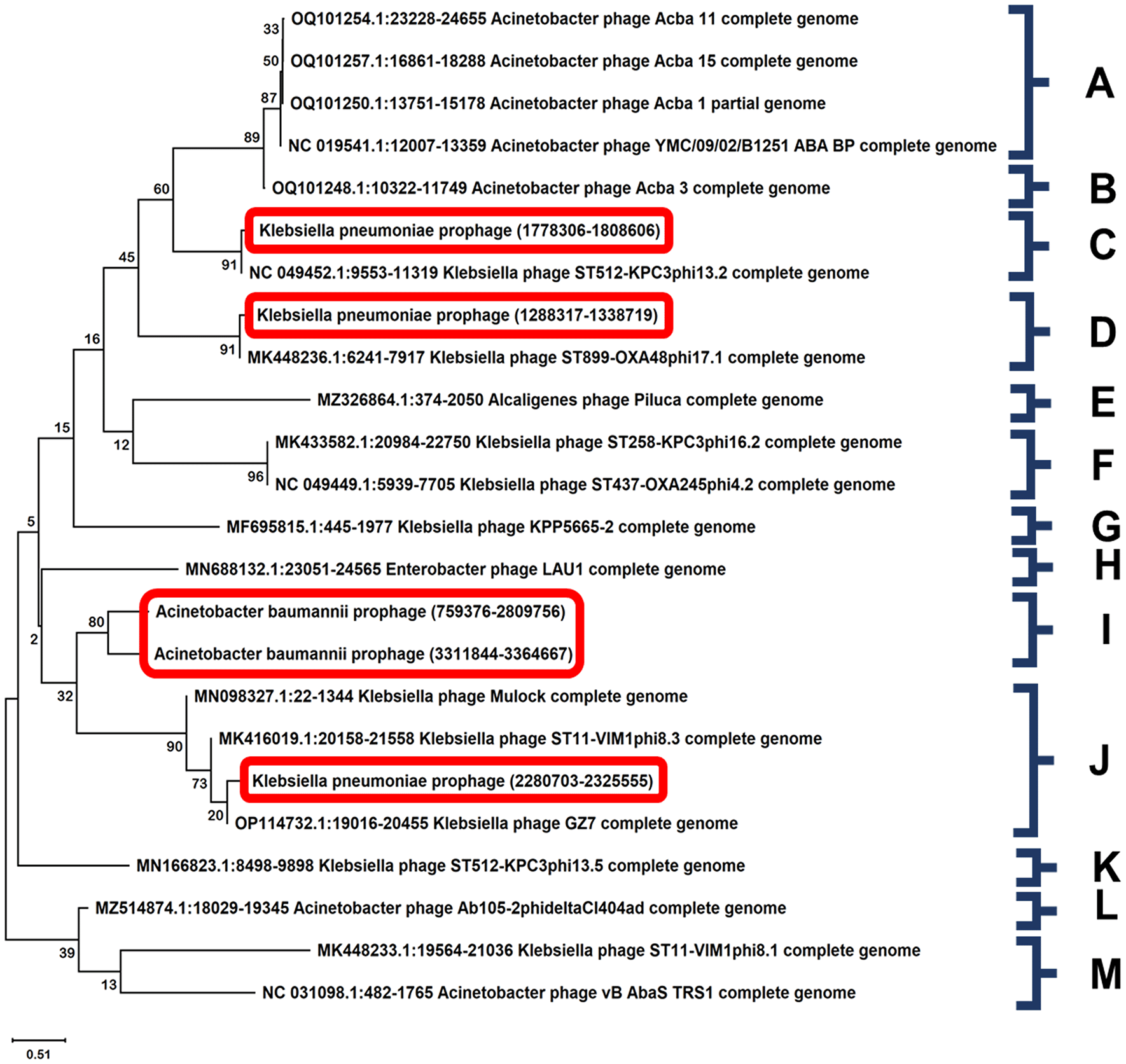
Phylogenetic analysis of predicted prophages was conducted based on their large terminase subunits. A neighbour-joining phylogenetic tree was generated with 1000 bootstrap replications using MEGA 11 (Figure 5). Phylogenetic analysis of the selected prophages along with the similar phages provided an insight into their diversity and relatedness. The Klebsiella pneumoniae prophages were present with other known Klebsiella pneumoniae phages showing close relatedness, whereas the prophages from Acinetobacter baumannii AB0057 sequences were closely related in a monophyletic order.
A prophage is a phage genome incorporated into the genome of bacteria that can infect and destroy a bacterial cell once it is expressed (Fortier and Sekulovic, 2013; Premetis et al., 2023). The presence of potentially useful and exploitable prophages within a bacterial genome can be detected and annotated via metagenomic data mining and bioinformatic algorithms and tools. Hence, this study considered the identification and annotation of prophage sequences that have been integrated into the genomes of MDR bacterial strains. After conducting rigorous screening based on annotations and sequence similarity analyses, we detected five prophages which met various selection criteria for an ideal infective prophage. From this selection, a cocktail of phages was computationally designed, consisting of phages that could collectively target the MDR strains of Acinetobacter baumannii and Klebsiella pneumoniae. Each phage in the cocktail is selected for its ability to target specific MDR strains of Acinetobacter baumannii and Klebsiella pneumoniae. By using a combination of phages, the intention is that the cocktail will have improved effectiveness as a treatment since it can target multiple bacterial strains and potentially reduce the probability of resistance development, thereby enhancing overall therapeutic efficacy.
Antibiotic resistance has propagated due to factors including the overuse or misuse of antibiotics, gene transfer, and inadequate diagnostics (World Health Organization, 2023; Mittal et al., 2020; Chinemerem et al., 2022; Ventola, 2015). Antibiotic-resistant bacteria like carbapenem-resistant Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa, have become an issue of global concern. These gram-negative bacteria are difficult to treat and the infections they cause often lead to increased morbidity and mortality rates (Breijyeh et al., 2020). Previous studies have considered the whole genome analysis of carbapenem-resistant Klebsiella pneumoniae genomes and MDR Acinetobacter baumannii; bacterial strains that can cause serious human health problems (Adams et al., 2008; Cienfuegos-Gallet et al., 2022). Phage therapy utilizes viruses which can attack and infect bacterial cells and can, therefore, operate as an effective substitute for antibiotics due to their high specificity and ability to target bacterial cells without harming human cells (Adams et al., 2008). Hence, bacteriophages are potential bactericidal agents; advantageous for the treatment of drug-resistant gram-negative bacteria (Breijyeh et al., 2020). Phage cocktails can be formulated by the combination of several phages that collectively possess an enhanced efficiency to lyse bacterial cells and can also be re-tailored if resistance develops (Abdulateef et al., 2023; Menor-Flores et al., 2022). This study considered the design of an effective phage cocktail against the MDR bacterial strains: Acinetobacter baumannii AB0057, Klebsiella pneumoniae HS11286, and Pseudomonas aeruginosa UCBPP-PA1, using a computational pipeline. Previously, therapeutic phage cocktails have been designed against Staphylococcus and Vibrio cholerae (Yen et al., 2017; Kornienko et al., 2020). Resistance to the antibiotic carbapenem is known to be dominant in the bacterial strains we have considered (Hamidian & Hall, 2011; Wen et al., 2018; Ranquet et al., 2001; Bi et al., 2015).
Previously, a number of integrative genetic elements including prophage sequences were identified in bacterial genomes using TIGER and Islander software, including 7, 3, and 1 prophage from the genomes of Klebsiella pneumoniae HS11286, Acinetobacter baumannii AB0057, and Pseudomonas aeruginosa UCBPP-PA1, respectively (Mageeney et al., 2020b). In the present study, we used the identified prophage sequences and considered their functional annotations. In addition, we examined prophage sequences from the selected strains using the PHASTER tool (Arndt et al., 2016) and this correlated with the prophage detection results of Mageeney et al. (2020b).
A range of bioinformatic packages provide a means to reveal phage genomic data; with PHASTER, Prophet, Virsorter, PhiSpy and Phigaro algorithms utilized for identifying phages in Lactobacillaceae (Happel et al., 2022). Using this approach, prophage sequences that were beyond the average size range for bacteriophages in the NCBI Caudovirales database (less than 11.6 kb) were detected (Happel et al., 2022). Those prophages were excluded from further consideration since it was improbable that they would perform their function properly. Also, phages with a size larger than 800 kb, much larger than the size of known phage genome sizes, were excluded (Happel et al., 2022). Similarly, vertically inherited caudophage sequences have previously been detected in enterobacterial genomes, whilst discarding phages with a size lower than 30 kb, considering them to be non-functional (Bobay et al., 2014). Hence, in this study, we selected only those prophages with a size greater than 30 kb for cocktail design, and none were larger than 60 kb; indicative of prophages that can code for complete and viable bacteriophages.
A total of 472 ORFs were predicted from the 11 prophages analyzed. We used the BLASTP algorithm to annotate the predicted ORFs in the prophage genomes. The NCBI non-redundant protein database which contains known protein sequences was then used to consider putative functions and potential biological roles of the encoded proteins. A total of 193 ORFs in 3 prophages of Acinetobacter baumannii were identified with maximum identity percentages of 100% and minimum identity percentages of 30.84%, 40.68%, and 42.82%, respectively. The Acinetobacter phage 5W, effective against MDR Acinetobacter baumannii (Peng et al., 2022), was found to share high sequence similarity with our putative prophages. About 265 ORFs in 7 prophages of Klebsiella pneumoniae were identified with maximum percentage identities of 100% and minimum identity percentages of 93.76%, 27.51%, 87.79%, 93.83%, 59.29%, 70.18%, and 86.84%, respectively. A BLAST analysis of Klebsiella prophages showed sequence similarity with the Klebsiella phage Mulock. The Klebsiella phage KPP5665-2 and Klebsiella phage Mulock have been previously used for bacteriophage therapy against resistant strains of Klebsiella (Herridge et al., 2020). A total of 14 ORFs were identified in the prophage of Pseudomonas aeruginosa which showed maximum identity of 100% and minimum identity of 38.22% in BLAST analyses. We found that Pseudomonas phage Pf1 was the most identical, which is known to be effective against other Pseudomonas strains (Peng et al., 2022).
Prokka annotation of these prophages enabled us to acknowledge their features including coding sequence (CDS) regions, ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs). This annotation provided insight into the diversity and development of these mobile genetic elements, as well as their possible interactions with bacterial hosts and other mobile genetic elements. We also identified putative promoter sequences and regions in the 11 prophage genomes. Our study revealed that three prophages of Acinetobacter baumannii had 47, 126, and 133 putative promoters, respectively. Among the 7 prophages of Klebsiella pneumoniae, the number of putative promoters ranged from 19 to 108. For the single prophage of Pseudomonas aeruginosa, we identified 19 putative promoters. These findings provide insights into the potential regulatory mechanisms of these prophages and their interactions with their host bacteria.
Virulent genes encode for virulence factors, crucial for the spread of bacterial infection and the potential initiation of disease. These factors also aid bacteria in the evasion of host defences, enhancing their pathogenicity (Abdulateef et al., 2023). Thus, in order to prevent bacteriophage-mediated transmission of these virulence genes, we used the Virulence Factor Database (VFDB) to detect them in our prophage genomic sequences. The results revealed that one prophage of Klebsiella pneumoniae harboured a peptidoglycan DD-metalloendopeptidase family protein and an undecaprenyl-phosphate glucose phosphotransferase, encoded by the wcaJ gene included in the cps gene cluster. This virulence factor is involved in the glycosyltransferase activity in the bacterial capsule (Paczosa & Mecsas, 2016). Additionally, the prophage of Pseudomonas aeruginosa was found to contain the phenazine biosynthesis protein PhzF, which is an isomerase that has been linked to virulence in P. aeruginosa (Blankenfeldt & Parsons, 2014). Thus, prophages in which these virulence genes were detected were rejected.
The Rho-independent transcriptional termination in bacteria involves the formation of a G/C rich 8-10 bp hairpin structure, followed by a uridine-rich stretch at the 3´ end of mRNA (Naville et al., 2011). This form of termination does not require the Rho factor and is adequate to trigger transcription arrest. This mechanism is characterized by sequence and structural signals within the genome, which can be identified via computational algorithms (Naville et al., 2011; Kingsford et al., 2007; Di Salvo et al., 2019). The detection of Rho-independent terminators is an important step in the annotation of phage genetic sequences as it characterizes the 3´ terminus of the mRNA transcript. We used ARNold program for the rapid identification of Rho-independent transcription terminators in our prophage sequences (Table 3).
The presence of 3 tRNAs encoded by 11 prophages was discovered through examination of the tRNA regions in the prophage genomes. Given that the host bacteria normally supply the tRNA molecules required for translation (Hudson et al., 2015), this result suggests that these prophages rely on the host translation machinery for protein synthesis. We also attempted to detect rRNA genes, which can interact with the host cell translation and protein production machinery. However, no rRNA genes were detected in the predicted prophages, but this does not have any effect on phage functioning because a bacteriophage hijacks the host translation machinery and replicates inside the host cell (Hamidian et al., 2019). A dot plot of selected phages was generated, which enabled us to reject K. pneumoniae prophage (4049881-4085225) which shared high sequence similarity with the K. pneumoniae prophage (1778306-1808606).
Small prophages with sizes below 11 kb, are considered to be non-functional and unable to assemble into a mature and viable bacteriophage, and only an intact phage with complete functional units can completely disrupt a target bacterium (Happel et al., 2022). The prophages with a size below 30 kb are also considered small, and it is difficult to differentiate between them and other mobile genetic elements (Bobay et al., 2014). This can pose a challenge when attempting to accurately identify and classify small prophages within prokaryotic genomes, therefore, we included only those prophages in our study which had a size greater than 30 kb.
A comparative genome analysis of the prophages was conducted using EasyFig and genome maps were created to consider the similarities between two identical genomes of prophages. The selected prophages with the highest similar phage sequences were analyzed using EasyFig 2.2. This provided a visualization of the similarities and differences between the prophages and their closest homologs. The identified prophage Acinetobacter baumannii (2759376-2809756) shared 96.57% sequence similarity with Acinetobacter phage YC#06; a virulent Acinetobacter phage YC#06 (accession number: ON391949.1) active against MDR Acinetobacter baumannii 4015, that was previously identified and characterized for formulating a phage-antibiotic combination therapy in vitro (Luo et al., 2022).
After performing BLASTn analyses, we reviewed available studies on phages having 100% and 99.9% similarity and a good query coverage with the prophages that we had detected and considered their ability to target any additional bacterial species or strains in addition to the aforementioned strains. We found that our selected prophage Acinetobacter baumannii 2759376-2809756 shared 96.06% identity with Podoviral Bacteriophage YMC/09/02/B1251 ABA BP (NC_01954.1) up to a query coverage of 40%. This bacteriophage belongs to the family Podoviridae and has a double-stranded circular DNA genome with a length of ~45kb and a 39.05% GC content. A complete analysis of Bacteriophage YMC/09/02/B1251 ABA-BP reported that this phage could lyse an isolate of the carbapenem-resistant Acinetobacter baumannii (CRAB) strain from a septic patient (Jeon et al., 2012). Previously, phages targeting a wide range of Klebsiella strains have also been reported, among which some of the phages were also included in our study. The phage (MK416019.1) was found to target K. pneumoniae ST11-VIM1 and phage (MK433582.1) was found to target K. pneumoniae ST258-KPC3. In the same way, phages (MN166823.1 and NC_049452.1) were reported to target K. pneumoniae ST512-KPC3 and phage (NC_049449.1) targets K. pneumoniae ST437-OXA245 (Bagińska et al., 2023; Lavigne et al., 2004). These Klebsiella strains are carbapenemase producing and can contribute to nosocomial infections, pneumonia, and urinary tract infections (Bleriot et al., 2020). These previously identified prophages shared a good percentage identity with our prophage sequences and this investigation suggested that the phage cocktail we have designed might target the above-mentioned bacterial strains as well, in addition to our selected MDR bacterial strains.
A study on the determination of morphology and biological properties of 12 viable phages against MDR Acinetobacter baumannii was carried out previously, and these were further selected to be used against MDR Acinetobacter baumannii (Bagińska et al., 2023). BLAST analysis revealed that their selected phages shared sequence similarity with the phages detected against Acinetobacter baumannii in our study. Another study involved genomic analysis of 40 Klebsiella prophages which were integrated into 16 strains of carbapenem-resistant Klebsiella pneumoniae (Bleriot et al., 2020). They also included prophages, with high similarity to the Klebsiella pneumoniae prophages reported in the current study, and these phages contain proteins mainly involved in the structure of virions, replications, the transcription of virions, and the regulation of the lysogenic and lytic cycles (Bleriot et al., 2020).
In this study, we have selected a total of 5 prophages for designing a phage cocktail, following rigorous screening (Table 7). Phylogenetic analysis of prophages isolated from Acinetobacter baumannii and Klebsiella pneumoniae was performed using a neighbor-joining method and the sequences were selected based on BLASTn analyses. Prophage sequences with the highest similarity were selected and a phylogenetic tree was constructed based on large terminase subunits of selected phages. Two critical tasks are carried out by the terminase large subunit during a viral replication cycle. Firstly, it acts as an ATP-powered molecular motor to help viral DNA move into empty capsids. Secondly, it also serves as an endonuclease, cleaving the viral DNA to start and stop packing activity (Bleriot et al., 2020). The phylogenetic analysis of selected prophages with other closely related prophages provided important insights into the diversity and relatedness of phages. The Klebsiella prophages were found to be present with other known Klebsiella prophages showing close relatedness, whereas the prophages from Acinetobacter baumannii genomes were found to be monophyletic. In this study, the highest similar sequences for Acinetobacter baumannii ranged from 99.91 to 75.38%, indicating a relatively high degree of diversity among the phages. For Klebsiella pneumoniae, a total of 25 of the most similar phages were selected, with similarities ranging from 99.99 to 78.95%: indicative of a relatively high diversity within the species.
Recently computational programs and tools have revealed unprecedented insights for the detection, functional annotation, and similarity analyses of potential prophages integrated within bacterial genomes (Breijyeh et al., 2020). The phages can be isolated and propagated relatively quickly and can be tailored to target specific strains or combinations of strains. The identified phages are highly specific in their action, which allows for targeted and personalized treatment. This reduces the risk of disrupting the action of beneficial bacteria within the body, unlike traditional antibiotics which can lead to collateral damage to the microbiome (Mu et al., 2021).
A phage cocktail against MDR Acinetobacter baumannii and Klebsiella pneumoniae was predicted computationally, which may prove useful to combat these bacterial strains that are resistant to a variety of drug treatments. Through extensive sequence similarity analyses, it was discovered that this phage cocktail might possess an ability to target other carbapenemase-producing Klebsiella pneumoniae strains as well, in addition to the initially selected MDR bacteria. As an alternative to conventional antimicrobial treatment, this highly specific phage cocktail could be developed in vitro and employed as a therapeutic agent for tackling certain MDR bacterial infections.
Conceptualization: RN; Data Curation: AH, MAA and ZH; Formal Analysis: AH and MAA; Project Administration: UM; Supervision: RN and MI; Validation: AH, MAA, MS, AR, and WGC; Visualization: RN and WGC; Writing – Original Draft Preparation: AH, MAA and AA; Writing – Review & Editing: HAR and WGC.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)