Keywords
Bioinformatics, Machine Learning, Type-2 Diabetes, Proteins, Pathways, Gene Ontology, RNA-Sequence, Drug
This article is included in the Machine Learning in Drug Discovery and Development collection.
Bioinformatics, Machine Learning, Type-2 Diabetes, Proteins, Pathways, Gene Ontology, RNA-Sequence, Drug
Differential Gene Expression (DGE) analysis usually based on the DESeq2 package1 is a traditional and common bioinformatics technique that helps to identify expressed genes under different conditions offering insights into genes that exhibit varying expression levels.2 In RNA sequence data, fold change in gene expression studies can be biased, potentially misclassifying genes with large absolute differences but small relative ratios.3 However, the advent of Machine Learning (ML) has brought about a significant change in bioinformatics, and it is now widely acknowledged as a powerful tool that can provide detailed and useful explanations of complex data that were once difficult to understand.4 And with the passage of time, in the medical sector, ML techniques are getting popularity, being effective for decision-making.5,6 Using different kinds of ML algorithms is noticeable in RNA sequence data for different types of detection and to find out the correlation of sequences,7 as well as for showing the effectiveness of machine learning algorithms in detecting splice variants from RNA sequence data.8 Such as: To identify and classify cancers early on, different computer algorithms have been used on microarray data sets. These include support vector machines, random forest, and neural networks.9 On the other hand, this study uses a neural network to analyze RNA sequence-expressed genes from different datasets to predict a patient’s health status.10 And in this paper, the primary objective is to classify or identify different types of cancers based on the patterns found in the gene expression data. By doing so, the research aims to enhance the accuracy and efficiency of cancer diagnosis, potentially leading to more targeted and effective treatments11 that inspired us to apply ML models in the bioinformatics field, especially in the RNA sequence count data.
Type 2 Diabetes (T2D), sometimes referred to simply as diabetes, is a long-term illness that affects the metabolic process.12 According to IDF, around 6.7 million people were dead in 2021, which is one of the major ten reasons for death in the universe, and around 541 million adults are affected by T2D.13 It was also projected in 2021 that by 2030, 643 million people would have diabetes, and by 2045, 783 million people will have the disease.14 However, the risk of serious complications from T2D is greatly reduced if it can be diagnosed in its early stages.15 Moreover, pioneers of improved biotechnology invented several bioinformatics tools that assisted the course of study about T2D.16 Yet, other groups of researchers have relied on machine learning (ML)-based aid systems for forecasting chronic illnesses.17–19 Researchers have suggested utilizing machine learning-based classification models to estimate the prevalence of T2D depending on its risk factors.20–23 So, this information encouraged us to be involved with T2D.
In our research on individuals with T2D, we utilized a feature importance method using XGBoost to identify highly expressed genes from RNA sequencing count data detecting T2D and not T2D individuals based on count data. This approach was used instead of relying solely on adjusted p-values and log fold Change values to determine significant genes. By training various algorithms on RNA sequence count data, we achieved notable prediction accuracies, with XGBoost emerging as a standout, and this approach not only enhances gene detection accuracy but also challenges traditional bioinformatics metrics, suggesting a richer machine learning-driven perspective on the genetic prospect of diseases like T2D. Moreover, on our detected expressed genes or significant features, we went through several bioinformatics analyses such as pathways, gene ontologies, protein-protein interactions, transcription factors, miRNAs, and drug predictions to deal with T2D. More importantly, we validated our findings with past studies to show the effectiveness of our models. So, in the future, this study will help researchers to gain knowledge more about mutated genes through machine learning, and to think about the prevention of T2D based on bioinformatics analysis like drug prediction discussed in this paper.
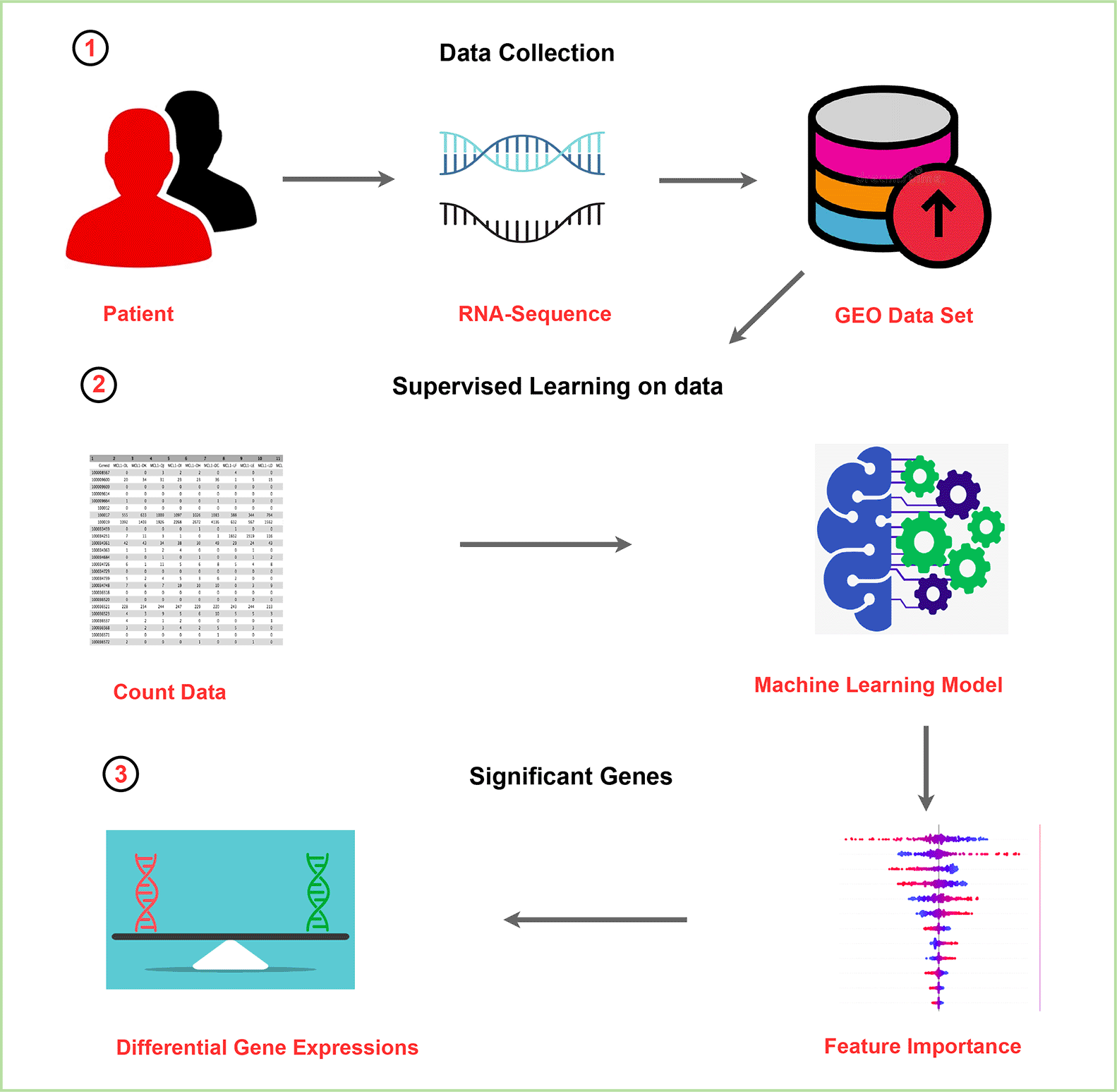
We initiated our study by selecting the RNA-Sequence count dataset (GSE81608)24 from GEO.25 This dataset was chosen for its reliability, especially in the context of biological data. The specifics of this dataset are detailed in Table 1. For ease of access and analysis, we downloaded the raw-count data from GREIN.26 Afterward, the data underwent a pre-processing through Utilizing the “pandas” library27 step to prepare this data for machine learning training.
On the preprocessed data, we trained six distinct ML models: Random Forest, AdaBoost, Gradient Boosting, Logistic Regression, Decision Tree, and XGBoost on the 80% of our dataset where 20% was for test. These models were trained on two classes: diabetes and non-diabetes. Our primary objective was to identify significant features28 which in our study were considered as differentially expressed genes.29 The entire machine learning process on this dataset is depicted in Figure 2. Additionally, for a more intuitive understanding, we visualized the RNA sequencing data and DGE in 4.
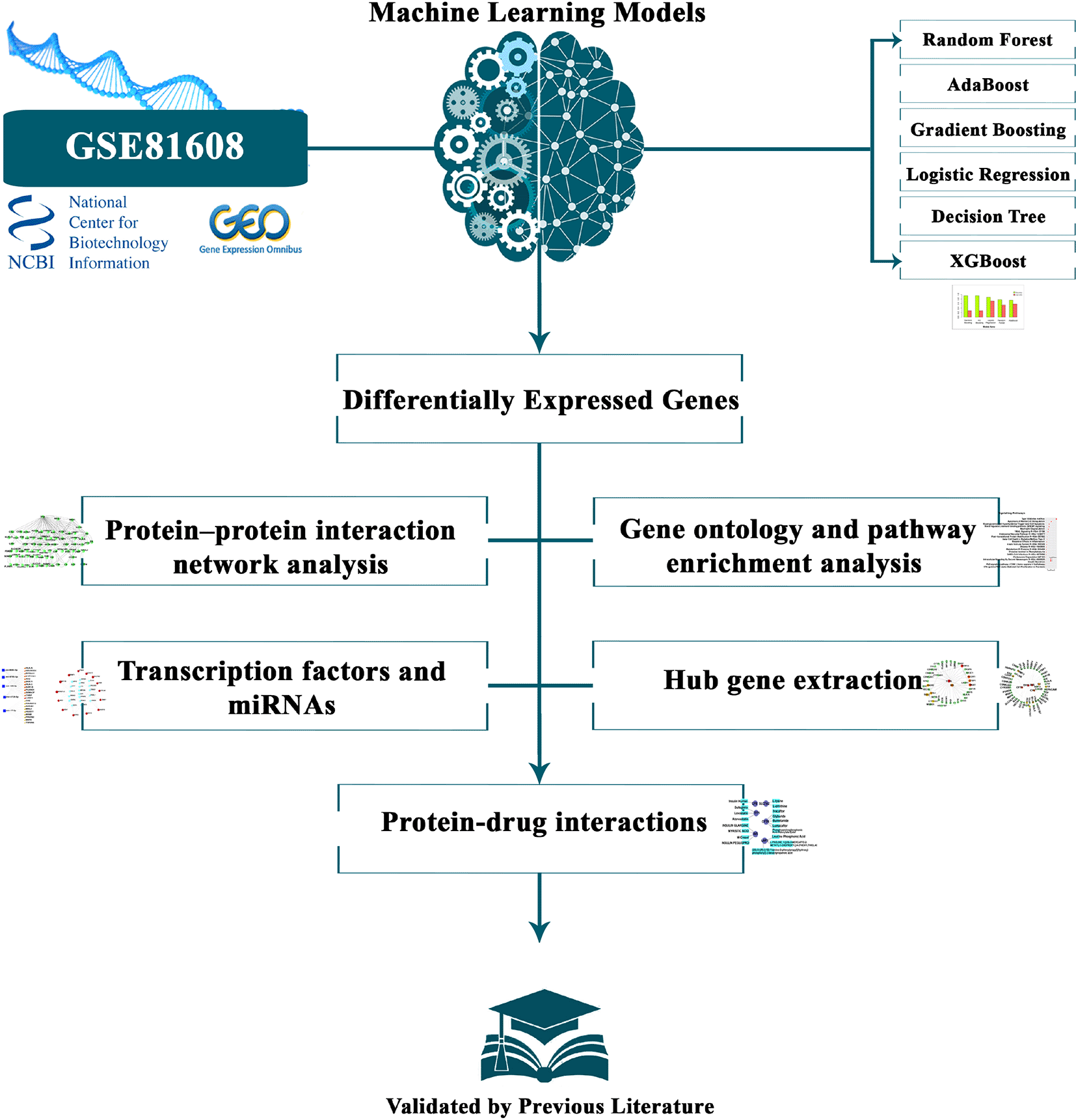
On our findings (significant features), we delved deeper into the data using a set of bioinformatics techniques. This comprehensive approach covered protein-protein interaction network analysis, gene ontology and pathway analysis, transcription factors and miRNAs analysis, hub gene extraction, and protein-drug interactions. To ensure the robustness and validity of our methods, we validated our results with existing literature. For a more intuitive understanding of our analytical approach, we have visually represented our methodology in Figure 1. For our gene ontology and pathway analysis, we turned to EnrichR.30 This tool provided insights into various sights of gene ontology, including biological processes, cellular components, and molecular functions. To further enrich our pathway analysis, we involved with information from trusted databases such as KEGG,31 Reactome,32 WikiPathways,33 Elsevier, and BioCarta.34 Throughout this process, we maintained an adjusted p-value of less than 0.05 as our benchmark for deciding significant pathways. On the other hand, our exploration into protein-protein interactions was simplified by the STRING online tool.35 Following this, we embarked on the creation of a hub-protein network, using the Cytoscape application with the cytohubba plugin.36 For insights into transcription factors, we recommended the JASPAR37 and ChEA38 databases. This allowed us to identify graphically plausible transcription factors that might connect with our differentially expressed genes (DEGs). This exploration was further enhanced using the NetworkAnalyst tool. Additionally, the TarBase39 and miRTarBase40 databases were instrumental in shedding light on miRNA-DEG interactions. NetworkAnalyst41 was the center of our analysis of TFs–gene and miRNAs–gene interaction networks. Moreover, for our exploration into drug-protein interactions, we relied on DrugBank,42 a comprehensive online resource of medicines and their associated drug targets.

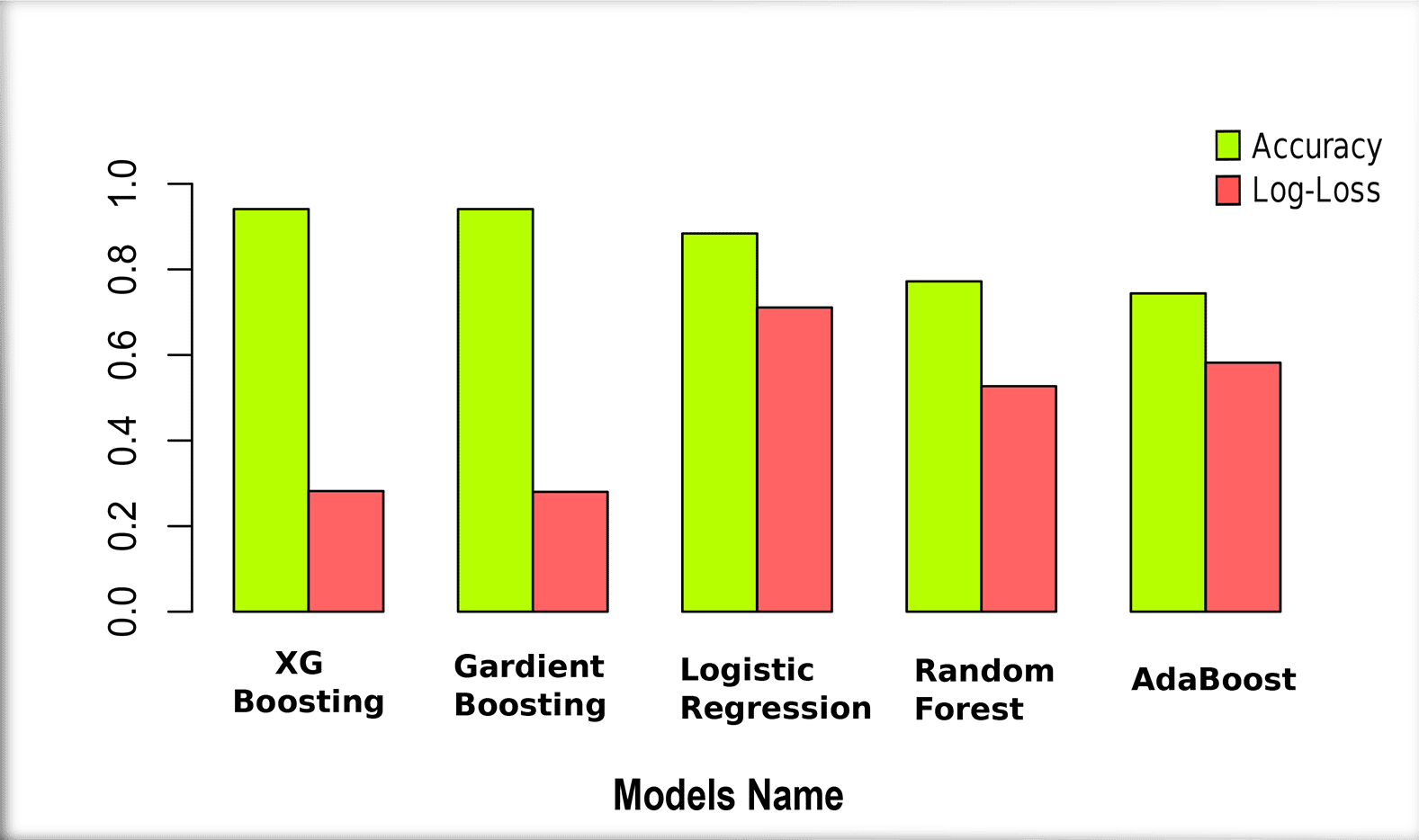
The evaluation of machine learning models is crucial across all domains but regardless of data related to biology is inescapable.79 While accuracy is a commonly used metric to evaluate a model’s performance, it alone may not provide a comprehensive assessment, especially in the context of biological or health-related datasets. Therefore, to achieve a clear understanding of a model’s efficacy, we incorporated a range of metrics including Precision,43 Recall,44 F1-Score,45 and Specificity,46 RocAuc, True Positive Rate, and False Positive Rate. These metrics collectively offer a perspective on the model’s performance, capturing various aspects of its predictive capabilities. The confusion matrix serves as a foundational tool in this context, covering the various measures employed to evaluate the efficiency of a classification model. However, To diagnose T2D from RNA sequence data, we used five unsupervised models Gradient Boosting, XGBoost, Logistic Regression, Random Forest, and AdaBoost. The XGBoost model performed well, with the highest prediction accuracy at 0.941% and the second lowest Log-Loss at 0.282%, showing values of confusion matrix for Precision 0.943, Recall 0.958, RocAuc 0.937, Specificity 0.915, F1-Score 0.950, TPR 0.958 and TFR 0.085%. Besides, the Gradient Boosting model showed the second highest prediction accuracy at 0.941% and the lowest Log-Loss at 0.280%, representing values of confusion matrix for Precision 0.948, Recall 0.943, RocAuc 0.937, Specificity 0.915, F1-Score 0.950, TPR 0.958 and TFR 0.085%. On the other hand, AdaBoost is the worst model in this accuracy, with the lowest accuracy of 0.744%. In addition, the accuracy is 0.884% in the third and 0.772% in the fourth positions for Logistic Regression and Random Forest, respectively. All the values have been shown in the Table 2, and the Figure 3 visualize the performance of the models based on the Accuracy and Log-Los.
Although we trained 5 different models to get our significant features (expressed genes), we selected the top 100 important genes based on our best-performed model, XGBoost with 0.941% accuracy, among them because this one has been trained well and showed the best performance (shown in Figure 3). In addition, we included our predicted expressed genes in the Extended data supplementary file-1). However, by the way of using this method, we ignored the conventional method, Differential Gene Expressions (DEGs). For the XGBoost model, the parameters we used have been provided in Table 3.
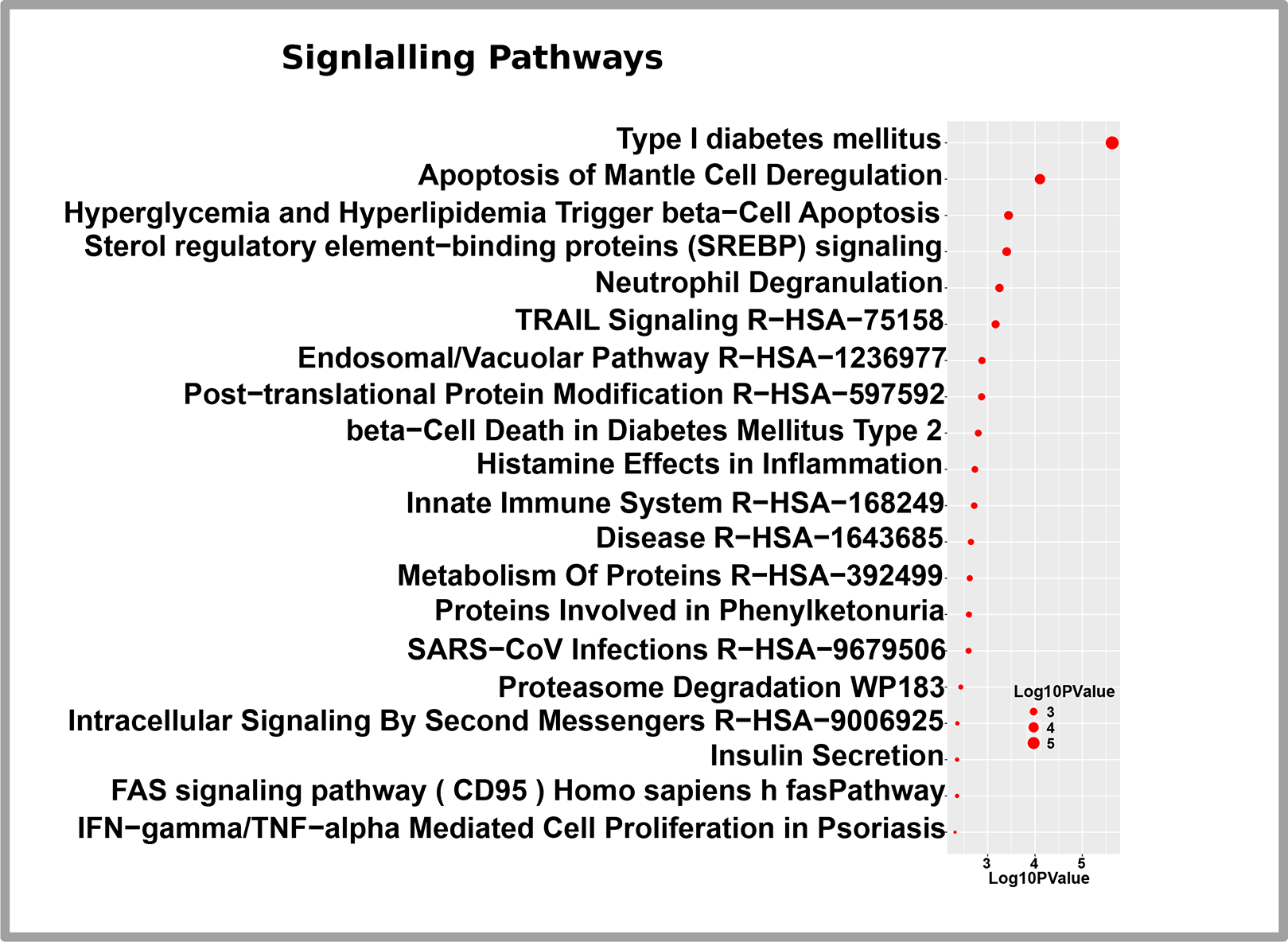
Utilizing the computational tool EnrichR, we conducted a gene set enrichment approach to determine pathways and took into account five pathways databases to conduct experiments utilizing DEGs of T2D. The 20 leading terms of signaling Pathways are presented in Figure 5. The top 10 terms in biological processes, molecular operations, and cellular components are included in Table 4. The adj. p-value, mostly less than 0.05, filters both the GO and the Pathways, which are then ordered ascendingly.
We used STRING to analyze the PPI network and a Cytoscape representation to predict the adherence pathways and recurrent interactions between DEGs. Utilizing topological metrics, such as a degree higher than 15°, extremely communicating proteins were defined via PPI interpretation. The most prominent DEGs include 75 nodes in this PPI network (shown in Figure 6) and 226 edges between them. Hub genes have a strong association in potential units and top 10% interconnectivity. Due to these interconnections, hub genes typically play a crucial role in biological systems. To find the top 18 DEGs (hub genes), we used Cytoscape’s Cytohubba plugin. Figure 7 illustrates the hub genes notably: TP53, INS, KDM1A, SNAI1, RCOR1, CTBP1, RPA1, RAD52, SQLE, CYP51A1, CFTR, CPE, C3, PRMT1, NFYB, CD38, CFP and CASP10. These identified hub proteins could be useful as therapeutic targets, yet their roles still need to be explored. T2D-related differentially expressed genes (DEGs) and their hub genes are summarized in Table 5.

The circular nodes in the diagram symbolize differentially expressed protein genes, while the edges depict the communication between nodes. The PPI consists of 75 nodes connected by 226 edges. The PPI network was generated utilizing STRING and visualized via Cytoscape.
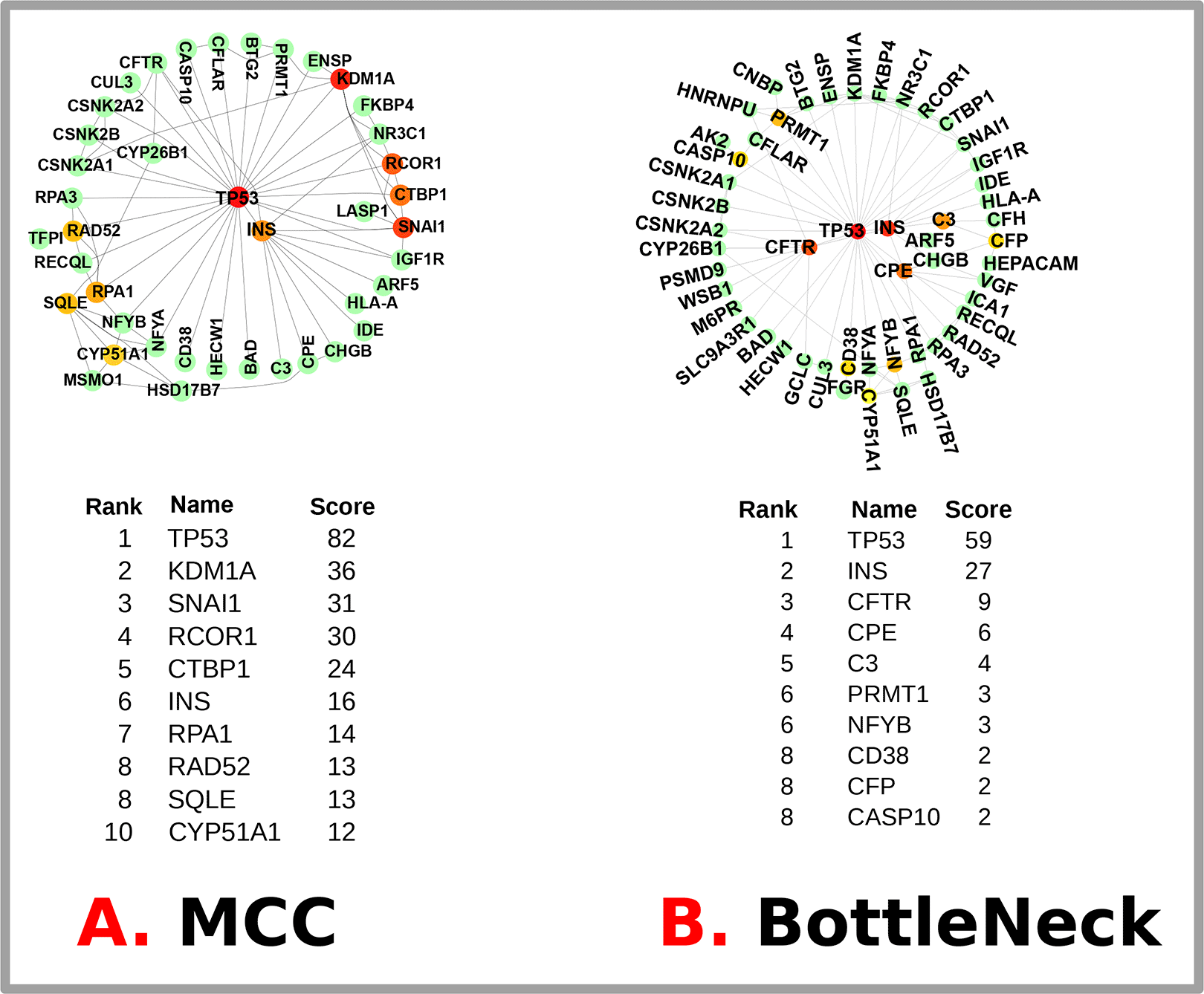
The most up-to-date MCC and BottleNeck techniques available in the Cytohubba plugin were used to obtain hub genes. The top 14 hub genes from each approach are highlighted below, along with the links between them and other compounds. BottleNeck contains 58 nodes and 100 edges, but the MCC network has only 48 nodes and 90 edges.
We used a network-based strategy to parse the governing TFs and miRNAs to locate substantial transcriptional changes and learn more about the hub protein’s signaling molecules. Transcription factors are proteins that govern gene activity and transcription over all life forms.47 Tiny RNA molecules called miRNAs have a role in post-transcriptional expression regulation. We investigated the interaction between DEGs and TFs, as shown in Figure 8, and DEGs and miRNAs, as shown in Figure 9. Major promoters of the TFs of differentially expressed genes were ELK4, FOXC1, FOXL1, GATA2, JUN, MEF2A, NFIC, NFKB1, POU2F2, PPARG, RELA, TEAD1, USF2, YY1, PRRX2, STAT3, TP53, E2F1, CREB1, NANOG, CREM, RUNX1, TP63, AR, HNF4A, POU5F1, SOX2, MITF, SPI1, MYC, FLI1, SUZ12, and EGR1. Mir-6883-5p, mir-6785-5p, mir-149-3p, mir-4728-5p, mir-17-5p, mir-210-3p, mir-374a-5p, mir-21-3p, mir-129-2-3p, mir-7-5p, mir-16-5p, mir-1-3p, mir-124-3p, mir-155-5p, mir-27a-3p, mir-34a-5p, let-7b-5p, and mir-107 were specified so that a concise overview of the DEGs operating at post-transcriptional regulators could be established. This Table 6 summarizes both transcriptional and post-transcriptional regulatory factors of type 2 diabetes-related differentially expressed genes.
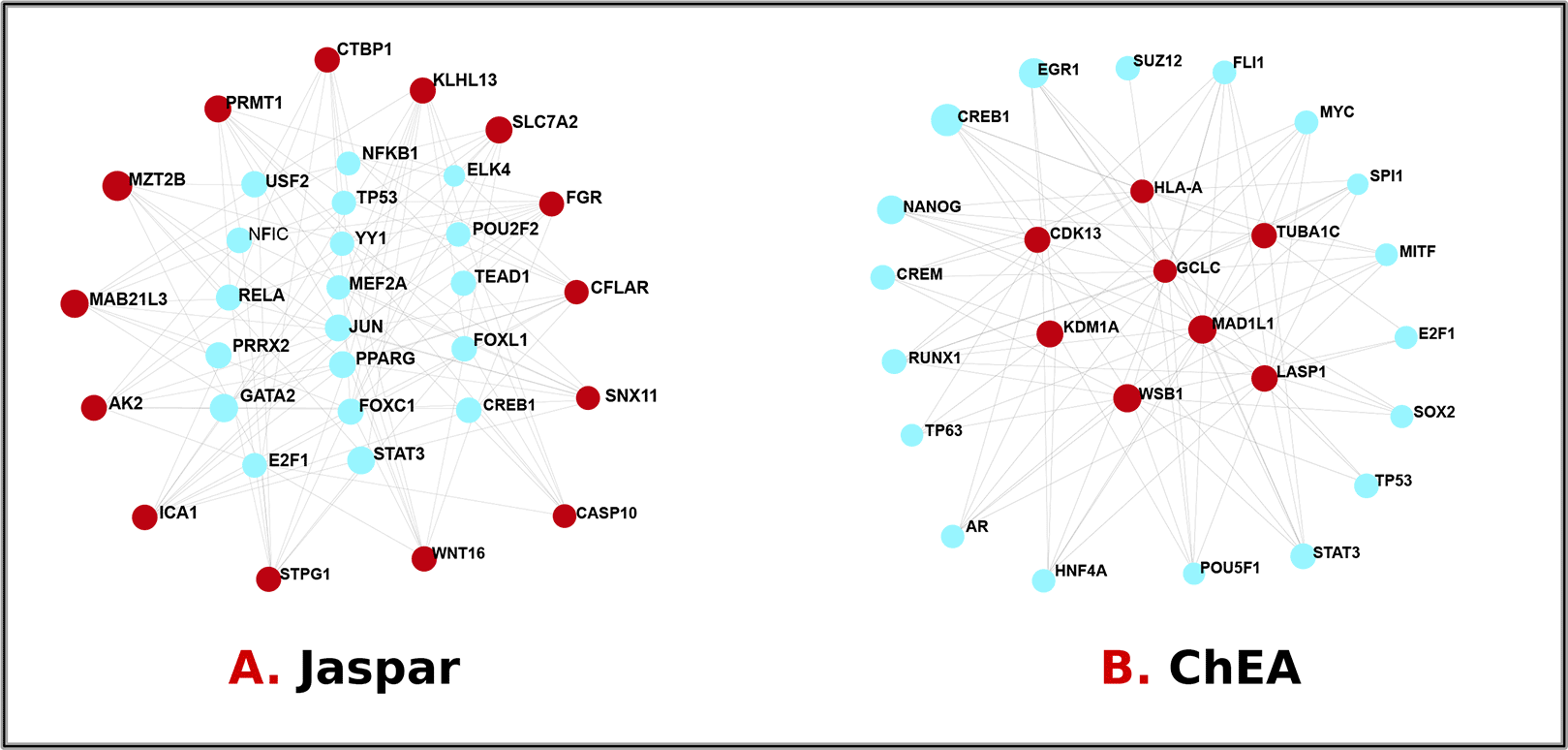
The circular cyan nodes represent transcription factors, while the circular red nodes represent gene icons that connect with transcription factors.

Here, the square node represents miRNAs, while the circular-shaped gene symbols connect with miRNAs.
To understand the structural features implicated in signal transduction, conducting a protein-drug interaction analysis48 is necessary. We listed 18 potential treatment drugs for frequent DEGs as possible pharmacological candidates in T2D employing NetworkAnalyst techniques dependent on drug-protein connections from the DrugBank library. Figure 10 shows 14 well-known therapeutic agents, including Insulin Human, Dalteparin, Lovastatin, Atorvastatin, Insulin glargine, Myristic acid, M-cresol, Insulin peglispro, L-lysine, L-ornithine, Ivacaftor, Glyburide, Bumetanide, and Lumacaftor that were found in the Protein Drug Interactions of DEGs of T2D. The potential uses of the remaining four chemical compounds in healthcare are still being investigated.
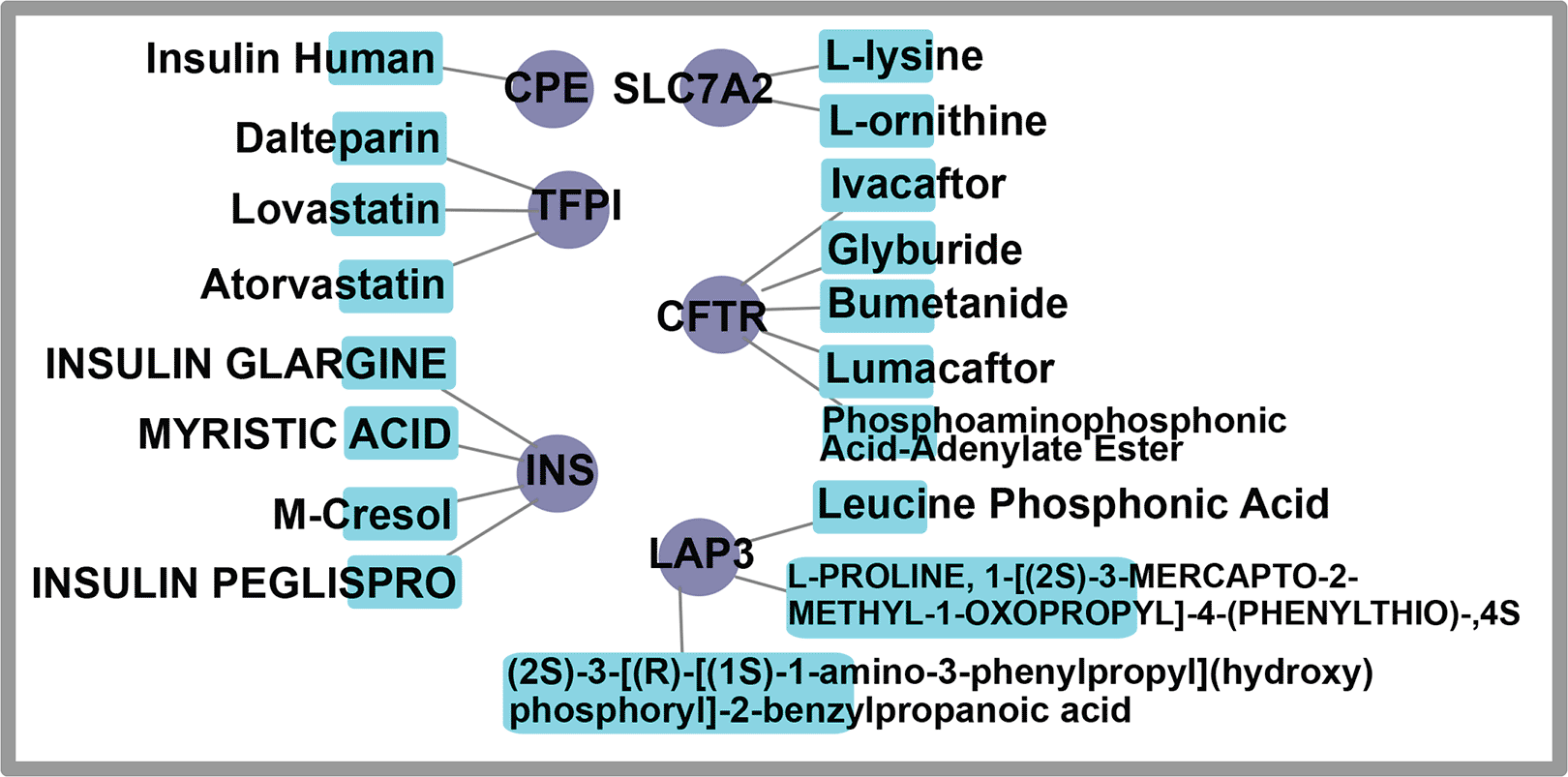
Here, the rectangular node symbolizes drugs, whereas the circular gene symbols are linked to drugs.
In the modern era, over time, as artificial intelligence is improving rapidly, Machine Learning is performing as an essential part in the bioinformatics sector analyzing data profoundly.49 Although we can use ML techniques on most of the RNA sequence data, in this research, we have analyzed T2D data because it is a chronic illness that can have severe and life-threatening complications.15 In this study, we presented a count-based classification pipeline to identify expressed genes applying the feature importance technique, as well as to detect the patient based on count data. Moreover, the approaches used here enable us to process large amounts of transcriptome data and draw reliable conclusions regarding T2D proteins involving various bioinformatics techniques, allowing us to comprehensively understand T2D and identify associated biomarkers.
In our comprehensive investigation of Type 2 Diabetes (T2D), we employed several supervised machine learning algorithms, including Random Forest, AdaBoost, Gradient Boosting, Logistic Regression, Decision Tree, and XGBoost. Their performance metrics, accuracies, and losses are visually represented in Figure 3 and detailed in Tab-2. From a bioinformatics perspective, we conducted Pathway enrichment analysis (Figure 5), Gene Ontology assessments (Table 4), Protein-Protein Interaction studies (Figure 6), and explored Hub-Protein interactions (Figure 7), Transcriptional Factor interactions (Figure 8), miRNA interactions (Figure 9), and drug-protein interactions (Figure 10). Each hub gene was meticulously detailed with its features in Table 5. Furthermore, we provided an in-depth overview of both transcriptional and post-transcriptional regulatory differentially expressed genes in Table 6. Our machine learning models’ efficacy in identifying significant genes from RNA sequence data sourced from NCBI for T2D is illustrated in Figure 4. For a holistic understanding of our research approach, we’ve outlined the entire methodology in Figure 1. Our dataset is comprehensively presented in Table 1, and the parameters of our top-performing model, XGBoost, are shown in Table 3.
In terms of our best model XGBoost, XGBoost’s superior performance on our dataset can be attributed to several factors. Its ability to model complex non-linear relationships, combined with built-in L1 and L2 regularization, makes it adept at handling high-dimensional data. Unique features such as internal handling of missing values, tree pruning, and efficient column block computation further enhance its efficiency. The model’s adaptability in hyperparameter tuning, resilience to outliers, and capability to capture feature interactions likely contributed to its edge. Additionally, the inherent nature of some datasets might align better with gradient-boosted trees, suggesting that our data’s underlying patterns were particularly suited for XGBoost. The mathematical equation of the aim and process of the XGBoost model is shown below:
Given a dataset with samples and features, the prediction of the model for the instance at the iteration is denoted as . The objective function to be optimized in XGBoost at each iteration is:
Here, is the number of leaves in the tree and is the score assigned to the leaf.
The optimal structure of the tree is found by minimizing:
Gene Ontology (GO) and pathway enrichment analysis is a widely used statistical method in bioinformatics that helps researchers to gain insights into the biological relevance of extensive gene sets. In T2D, persistent exposure to high glucose levels and free fatty acids induces beta-cell dysfunction and may initiate beta-cell apoptosis.50 Sterol regulatory element binding proteins (SREBPs) regulate lipid production and adipogenesis. SREBPs expression was considerably reduced in individuals with type 2 diabetes.51 Neutrophil degranulation is related to an aberrant echocardiographic patterning T2D.52 The expression of the FAS signaling pathway (CD95) was connected to systemic and skeletal muscle insulin resistance.53 IFN-gamma or TNF-alpha Mediated Cell Proliferation is associated with T2D. Interferon-gamma is crucial for the ruination of cells and the onset of T2D.54 Tumor necrosis factor (TNF)-alpha, a cytokine derived primarily from macrophages and adipocytes, can encourage insulin resistance (IR) and inevitably aid the advancement of T2D (T2D).55 The innate immune system plays a crucial part in T2D by contributing to low-grade inflammation and insulin resistance, which are key factors in the development and progression of the disease.56 Several MHC class I alleles, such as HLA-B and MHC class II alleles(including HLA-DRB1 and HLA-DQB1) were associated with T2D risk.57,58
We identified hub proteins that are expressed highly or poorly in T2D patients. Patients with T2D had significantly greater serum TP53 levels than healthy non-diabetic controls.59 The decreased level of CTBP1-AS2 was linked with diabetes in the Iranian population.60 The presence of complement factor 3 (C3) is linked to insulin resistance.61 ROS- and RAS-mediated diabetic retinopathy involves PRMT-1 and DDAHs-induced ADMA upregulation.62 In a sizeable portion (9–15%) of patients with Type 2 or persistent Type 1 diabetes, CD38 autoantibodies have been discovered. Most of these autoantibodies (about 60%) exhibit agonistic characteristics, such as Ca2+ mobilization in lymphocytic cell lines and in pancreatic islets, indicating that they are biologically active. CD38 autoantibodies promote glucose-mediated insulin secretion in human pancreatic islets.63 The hypomethylation of CASP10 may result from T2D and severe and long-lasting hyperglycemia.64 In humans, CFTR deficiency causes intrinsic abnormalities in insulin secretion inside the islets.65
Our discovered TFs are associated with T2D. The adult human pancreas has been found to express the Sox2 gene. It seems improbable that Sox2 will have a genetic influence on the development of T2D.66 In the human intestine, MYC transcription factor expression is correlated with either glycaemic management (HbA1c level) or body mass index (BMI).67 In the skeletal muscle of T2D patients, STAT3 is constitutively phosphorylated, and increased STAT3 signaling plays a role in the etiology of T2D and insulin resistance.68 Much research has been conducted on the peroxisome proliferator-activated receptor gamma (PPARG), whose ligands have become effective insulin sensitizers in type 2 diabetes.69 YY1 plays an important role in T2DM and it might be useful as a new therapeutic target in the fight against the disease.70
Our identified miRNAs are also linked to T2D. Targeting the SOCS1-mediated NF-B Pathway, miR-210-3p Increases Insulin Resistance and Obesity-Induced Adipose Tissue Inflammation. MiR-374a-5p appears to be associated with the downregulation of pro-inflammatory biomarkers that are connected to insulin resistance and is elevated in metabolically healthy obese persons as compared to metabolically abnormal obese patients.71 A possible biomarker for the early diagnosis of diabetic nephropathy is miR-21-3p, whose expression is downregulated in association with the onset of diabetic nephropathy.72 MiR-7-5p targeting may be a likely therapeutic approach to metabolic illnesses brought on by insulin dysfunction.73 Patients with diabetic neuropathy have dysregulated long non-coding miR-1-3p axis.74 Compared to healthy individuals, peripheral blood mononuclear cells (PBMCs) obtained from T2D patients exhibited low miR-155 expression.75
Our identified potential drug molecules can be effective for T2D patients. Insulin Human N (medium-acting) and Insulin Human R (short-acting) both are used in diabetes mellitus to lower blood glucose levels. Insulin glargine modulates carbohydrate, protein, and lipid metabolism by suppressing hepatic glucose synthesis and lipolysis and improving peripheral glucose clearance. Long-acting insulin, or insulin peglispro, is used to treat both T1D and T2D.76 Glyburide belongs to the class of medications known as sulfonylureas, which stimulate the pancreas to produce insulin and reduce blood sugar levels.77 T2D and its associated illnesses may be prevented and treated using myristic acid.78 There is some uncertainty on how Dalteparin, Lovastatin, Atorvastatin, Myristic acid, M-cresol, L-lysine, L-ornithine, Ivacaftor, Bumetanide, and Lumacaftor interact with T2D. So, more studies as well as preclinical and clinical trials are required.
As our research aims to provide the machine learning approach to identify significant genes through feature importance technique and to address the global burden of T2D and enhance the lives of individuals, affected by this chronic disease, based on bioinformatics methods, we went through machine learning approach and common bioinformatics methods.
In terms of the machine learning approach, our research represents a remarkable advancement in bioinformatics and (T2D) research. Combining machine learning algorithms, statistical analysis, and bioinformatics techniques, we have gained valuable insights into the molecular mechanisms underlying T2D. It would help us to identify expressed genes not solely being dependable on the conventional bioinformatics approach, Differentially Expressed Genes (DEGs). As well as, for a large number of features, a machine learning approach would be an effective and more reliable approach to predict expressed genes. We have successfully processed and analyzed vast amounts of information by utilizing large volumes of transcriptomic data. Our XGBoost model could be an example that showed a noticeable performance with 94.1% accuracy. This achievement demonstrates the potential of machine learning as a powerful tool for precise and efficient T2D diagnosis, which also can significantly impact clinical practice by enabling early intervention and personalized treatment approaches based on count data.
On the other hand, in our comprehensive molecular biomarker study on T2D, we analyzed a diverse set of molecular entities, including 20 pathways, 30 gene ontologies, 51 transcriptional factors, 18 hub-genes, 18 miRNAs, and 18 potential drugs where we were able to validate a subset among them. So, according to our validation, key pathways such as SREBPs, neutrophil degranulation, FAS signaling, IFNgamma/TNFalpha-mediated cell proliferation, and the innate immune system were highlighted as central to T2D’s development. Hub proteins, notably TP53, CTBP1-AS2, C3, PRMT-1, DDAHs, CD38 autoantibodies, CASP10, and CFTR, present promising avenues for disease research and treatment. Similarly, transcriptional factors like Sox2, MYC, STAT3, PPARG, and YY1, coupled with miRNAs such as miR-210-3p, miR-374a-5p, miR-21-3p, miR-7-5p, and miR-155, shed light on the regulatory dynamics underpinning T2D. Furthermore, our research identified potential drug molecules, including insulin analogs, sulfonylureas, myristic acid, and other compounds, that hold promise for therapeutic intervention. While further preclinical and clinical trials are necessary to validate their efficacy and safety profiles, these findings offer potential avenues for revolutionizing T2D treatment strategies and improving patient outcomes.
Overall, this study represents a significant advancement in using machine learning to identify expressed genes from RNA sequence count data, and by integrating key bioinformatics methods on T2D and validating our findings against prior research, we offer a robust approach to understanding and addressing T2D at the molecular level.
One of the limitations of our study is the accuracy of our models, which needs further improvement. While our current approach provided insights, the application of deep learning models in future studies could enhance the precision of our findings. Additionally, there remains a set of biomarkers that we have yet to validate with existing literature. A thorough validation of these biomarkers is essential, as it would offer researchers a more detailed understanding of expressed genes, ultimately aiding in more accurate analyses and better strategies to address T2D.
Md Al Amin: Took the lead in conceptualization and design of the work; primary role in data curation and analysis; led the writing of the original draft of the manuscript; major contributor in review and editing; gave final approval of the version to be published; agreed to be accountable for all aspects of the work.
Feroza Naznin: Played a significant role in the formal analysis of the data; contributed to the investigation; involved in writing, review, and editing of the manuscript; gave final approval and agreed to be accountable for all aspects of the work.
Most Nilufa Yeasmin: Led the acquisition of resources; involved in data curation; gave final approval and agreed to be accountable for all aspects of the work.
Md Sumon Sarkar: Took the lead in software development; contributed to the validation of the results; gave final approval and agreed to be accountable for all aspects of the work.
Md Misor Mia: Led the visualization process; supported the investigation; gave final approval and agreed to be accountable for all aspects of the work.
Abdullahi Chowdhury: Contributed to the methodology; supported the writing, review, and editing of the manuscript; gave final approval and agreed to be accountable for all aspects of the work.
Md Zahidul Islam: Acted as the primary supervisor and administrator of the project; took a leading role in writing, reviewing, and editing the manuscript; gave final approval and agreed.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)