Keywords
Kabuki syndrome, mutation, exome, genetic study, intellectual disability, case report
Kabuki syndrome, mutation, exome, genetic study, intellectual disability, case report
Kabuki syndrome is a rare poly-malformative syndrome associated with variable degrees of intellectual disability and specific facial dysmorphology. It was first described by Niikawa et al.1 and Kuroki et al.2 in Japan in 1981. Kabuki syndrome is defined by five essential diagnostic criteria: specific craniofacial dysmorphology, growth retardation, intellectual disability, skeletal manifestations, and dermatoglyphic anomalies. Other clinical symptoms may also be present but are not always observed. The prevalence of Kabuki syndrome is estimated to be around 1 in 32,000 births.1 There are two genes responsible for this condition: primarily, germline heterozygous loss of function mutations in KMT2D gene, also known as lysine methyltransferase 2D (MLL2), which encodes a methyltransferase, and secondarily, the lysine demethylase 6A (KDM6A) gene, the second gene identified in this disorder, which codes for a histone demethylase. The number of individuals with mutations in this gene is currently low. In this case report, we present a case of Kabuki syndrome suspected in the prenatal period and confirmed by genetic analysis, revealing a novel mutation with a favorable prognosis, particularly regarding intellectual function. The objective of our report was to further elucidate the clinical and biological manifestations of this condition and highlight the importance of early genetic diagnosis.
We present the case of a nine-and-a-half-year-old boy who has been followed since birth for Kabuki syndrome. His mother has no significant medical history, and no similar cases have been reported in the family.
The mother was twenty-nine years old, Para 0, Gravida 1, with no familial history of genetic abnormalities or intellectual defect. She was referred by a private gynecologist to the Department of Gynecology and Obstetrics at Ben Arous Regional Hospital following the discovery at 26 weeks’ gestation of ventriculomegaly with bilateral pyelectasis. The pregnancy follow-up was irregular, and the mother didn’t undergo first-trimester combined screening for chromosome abnormalities.
Ultrasound performed at our department showed bilateral colpocephaly associated with an enlarged cavum pellucidum (Figure 1). Furthermore, biometric measurements did not reveal intrauterine fetal growth restriction. All tests for common infectious diseases such as Toxoplasmosis, syphilis, varicella-zoster virus, parvovirus B19, Rubella, Cytomegalovirus, and Herpes simplex virus come back negative.

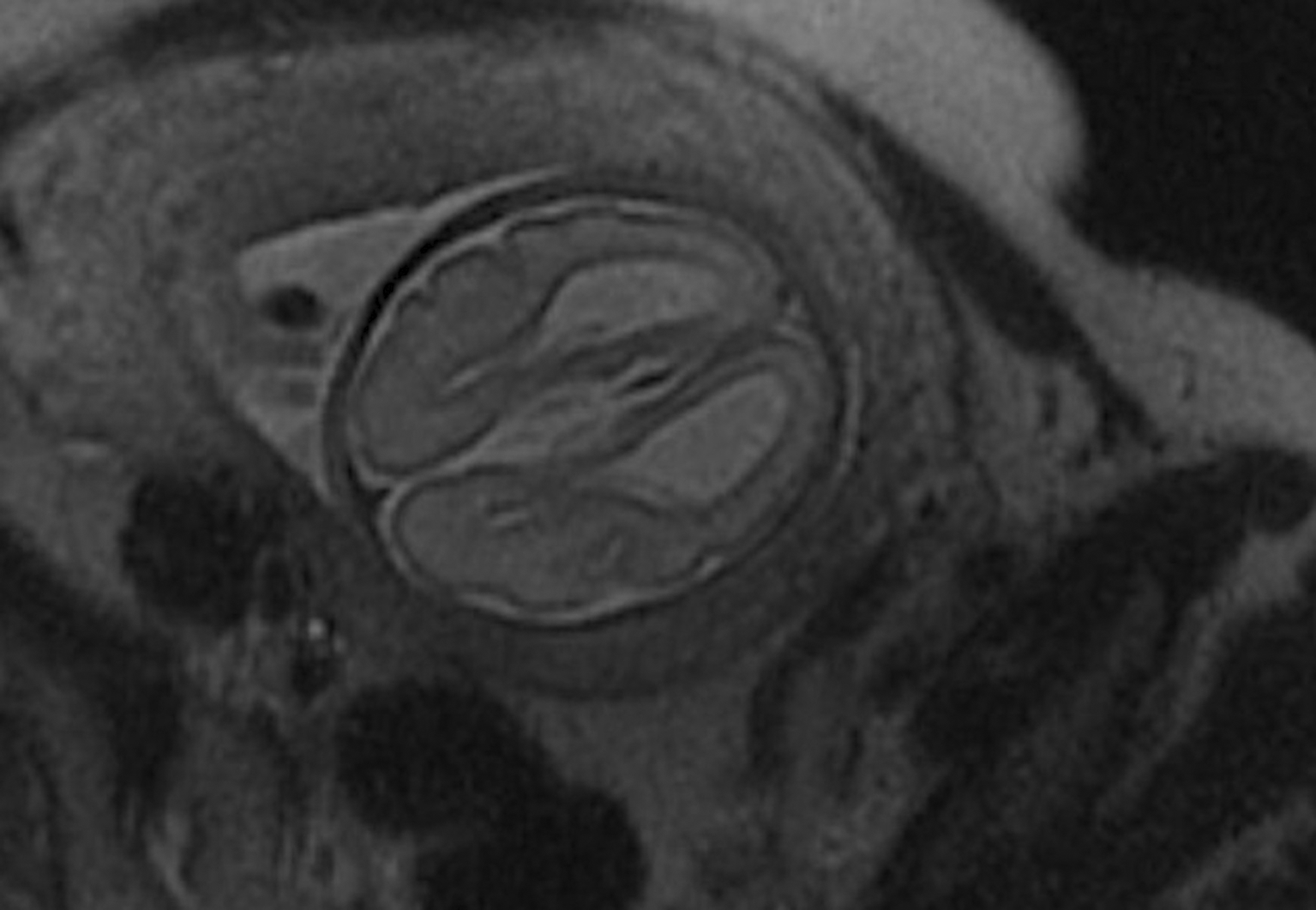
A fetal Magnetic Resonance Imaging (MRI) was performed at 28 weeks’ gestation, confirming the diagnosis of agenesis of the corpus callosum associated with ventriculomegaly, without other associated malformations, particularly no other abnormalities to the brain (Figure 2).
An amniocentesis was performed after parental consent to obtain standard karyotype, which showed a normal male 46, XY chromosomal formula.
A meeting was organized with the parents, involving gynecologists and pediatricians, to discuss the possible origins of the anomalies and their associated prognosis. The parents decided to continue the pregnancy to term without any further investigations.
The patient was born vaginally at 37 weeks’ gestation with good adaptation to extrauterine life. Upon initial examination, he was eutrophic with a weight of 3050 g, length of 49 cm, and head circumference of 34 cm. He exhibited facial dysmorphology, including a flat occiput, prominence of the metopic suture, an oval-shaped face with a narrow forehead, large almond-shaped eyes, and intermittent convergent strabismus of the left eye. Examination of the limbs revealed flat feet, without any other musculoskeletal malformations. However, upon examination of the external genitalia, no gonads could be palpated.
He was initially hospitalized for transient respiratory distress, which resolved within 48 hours. A grade 3 murmur was also detected upon cardiac auscultation at the aortic area. Upon examination of the external genitalia, gonads were not palpable. The neurological examination was unremarkable.
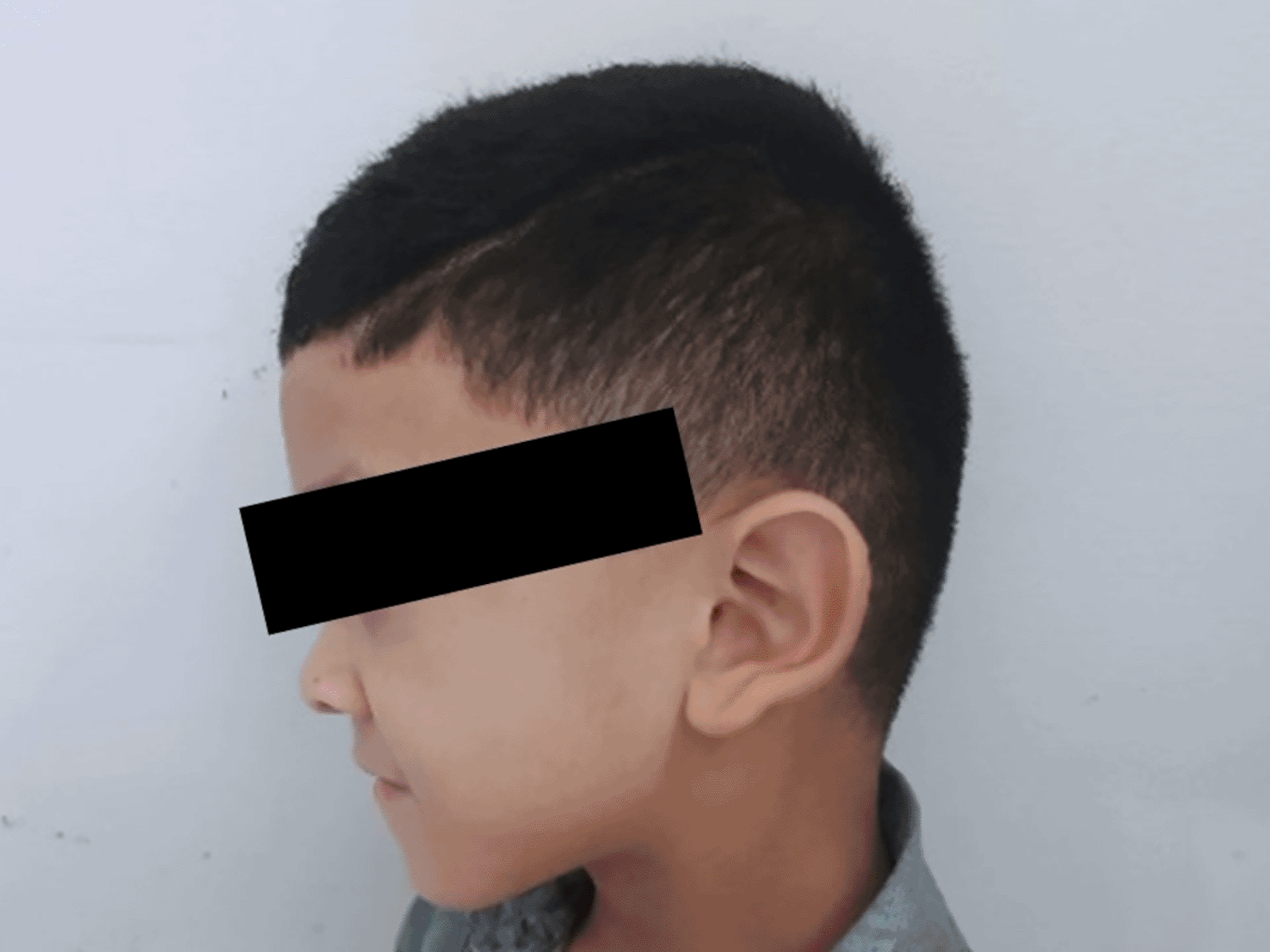
The patient’s facial dysmorphology became more evident at the age of 4 years and 9 months. It was characterized by a flattening of the occiput, marked microcephaly at (-2) standard deviations (SD) with prominence of the metopic suture, and an oval-shaped face with a narrow forehead. The eyes showed sparse outer halves of both eyebrows, increased eye size with heavy eyeliner, intermittent convergent strabismus of the left eye, inversion, or eversion of the lower eyelids on both sides, forward-tilted nostrils, a mildly marked philtrum with notable depression at the root and tip of the nose (Figure 3). The ears were wide and protruding, with outwardly turned earlobes. In the oral cavity, there was a high-arched palate, a small mouth with a thin upper lip, and micrognathia (Figure 3). In the limbs, bilateral brachydactyly of the hands was observed (Figure 4a) but radiological examination did not provide evidence supporting this finding (Figure 5). Hypoplastic nails of the 5th fingers and toes, bilateral persistence of fetal fingerprints, and ligamentous hyperlaxity with loose skin were also noted (Figure 4 a,b).

a: Bilateral brachydactyly of the hands; b: Hypoplastic nails of the 5th fingers and toes, bilateral persistence of fetal fingerprints, and ligamentous hyperlaxity with loose skin.

In terms of psychomotor development, there was a slight delay in achieving a sitting position at nine months and walking at eighteen months. Speech therapy evaluation revealed nasal speech with a slight delay in language acquisition, with the patient starting to form short phrases at the age of three.
Neurological investigations showed agenesis of the corpus callosum on transfontanellar ultrasound performed on day 32 of life. Given these lesions and the flat occiput, the diagnosis of Crouzon syndrome was suspected, which also remains a differential diagnosis of Kabuki syndrome. A subsequent brain CT-scan revealed a complete agenesis of the corpus callosum, associated with dilation and enlargement of the occipital horns of the lateral ventricles, colpocephaly, distortion of the frontal horns of the lateral ventricles resembling the appearance of bull horns, and ascent of the roof of the third ventricle. Partial filling and hypoplasia of the maxillary sinuses and agenesis of the frontal sinuses were also observed, with no signs of craniofacial dysostosis. A brain Magnetic Resonance Imaging confirmed the total agenesis of the corpus callosum, with an eccentric appearance of the lateral ventricles and bilateral dilations of the occipital and temporal horns.
Regarding neurosensory evaluations, an ophthalmological examination performed under general anesthesia revealed visual acuity of 5/10 with correction in the right eye and 7/10 in the left eye, along with convergent strabismus in the left eye. Auditory-evoked potentials were found to be normal.
On the therapeutic level, speech therapy intervention was carried out, and the following results were observed after rehabilitation: Currently, the patient presents with logorrhea, some pronunciation difficulties, and a normal IQ. He presents a significant improvement in communication, good comprehension of simple instructions, good acquisition of oral and written language, and no contraindications for school inclusion. The recommended approach and therapeutic plan included parental initiation and guidance, referral to a specialized center or other specialties for school integration, continued speech therapy, and recommended consultations with child psychiatry.
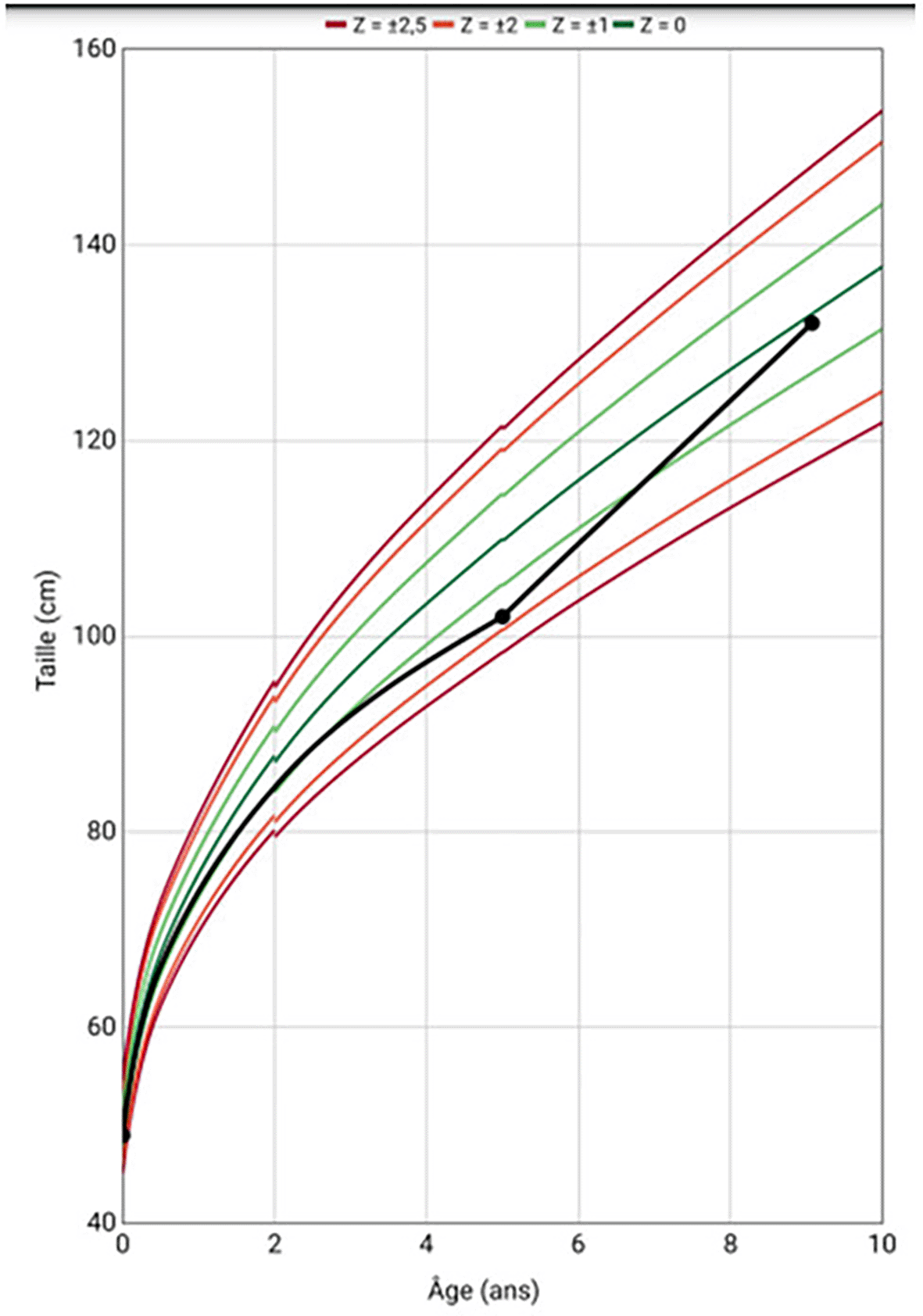
In terms of growth and weight, the patient experienced stature growth retardation with a deviation from the growth curve at the age of 5 years (Figure 6). Bone age evaluation performed at the chronological age of 5 years revealed a 2-year lag compared to the chronological age, suggesting a possible endocrine cause. Blood tests showed Thyroid-stimulating hormone (TSH) at 1.88 μIU/ml (0.35-4.94 μIU/ml), Thyroxin (FT4) at 13.97 pmol/l (9-19 pmol/l), cortisol level at 140 ng/ml (100-240 ng/ml), Adrenocorticotropic hormone (ACTH) at 28.6 ng/l (7.2-63.3 ng/l), Insulin Growth factor 1 (IGF1) at 87.53 ng/ml (associated with normal liver function), insulin stimulation test for Growth hormone (GH) secretion at 4.63 ng/ml (>20 ng/ml). The diagnosis of growth hormone deficiency was established. A Magnetic Resonance Imaging (MRI) of the hypothalamus-pituitary region showed no abnormalities. The patient was started on injectable growth hormone therapy at a dose of 0.030 mg/kg/day subcutaneously, with good progress (average height gain of 7.5 cm per year) (Figure 6).
Regarding the respiratory tract, the patient had recurrent episodes of bronchiolitis associated with pneumonia, with a frequency of two infections per year. The latest episode was at the age of eight years old. An initial immunodeficiency workup returned normal.
Regarding the cardiac aspect, the cardiac ultrasound performed at birth revealed a restrictive subaortic ventricular septal defect without significant impact on the heart. Subsequent echocardiographic evaluations showed the same aspect of the ventricular septal defect (VSD), and periodic monitoring was recommended without the need for surgical intervention. Currently, he presents with a small VSD without any significant impact.
The anomalies identified during the prenatal ultrasound have been confirmed. An abdominal ultrasound performed at the age of one month revealed left renal megacalycosis. A follow-up ultrasound at the age of one year showed left pyelectasis with anterior-posterior pyelic diameter (APPD) of 16mm, without visible obstruction, consistent with pyelo-ureteral junction syndrome. A renal scintigraphy performed at the age of 4 years showed left pyelectasis with satisfactory non-obstructive drainage and symmetrical renal function. The right kidney showed early perfusion and uptake with delayed drainage, responding fully to Lasix injection, with an estimated relative function of 48%. The left kidney showed normal perfusion and uptake, with pyelectasis and delayed drainage responding to Lasix injection, with an estimated relative function of 52%. A retrograde uretero-cystography was performed, and showed no abnormalities, particularly no passive or active vesicoureteral reflux. The patient is currently being followed up in pediatric surgery. He did not experience urinary tract infections.
Additionally, in the presence of empty scrotum, testicular ultrasound revealed two inguinal testicles. He underwent surgery without any complications.
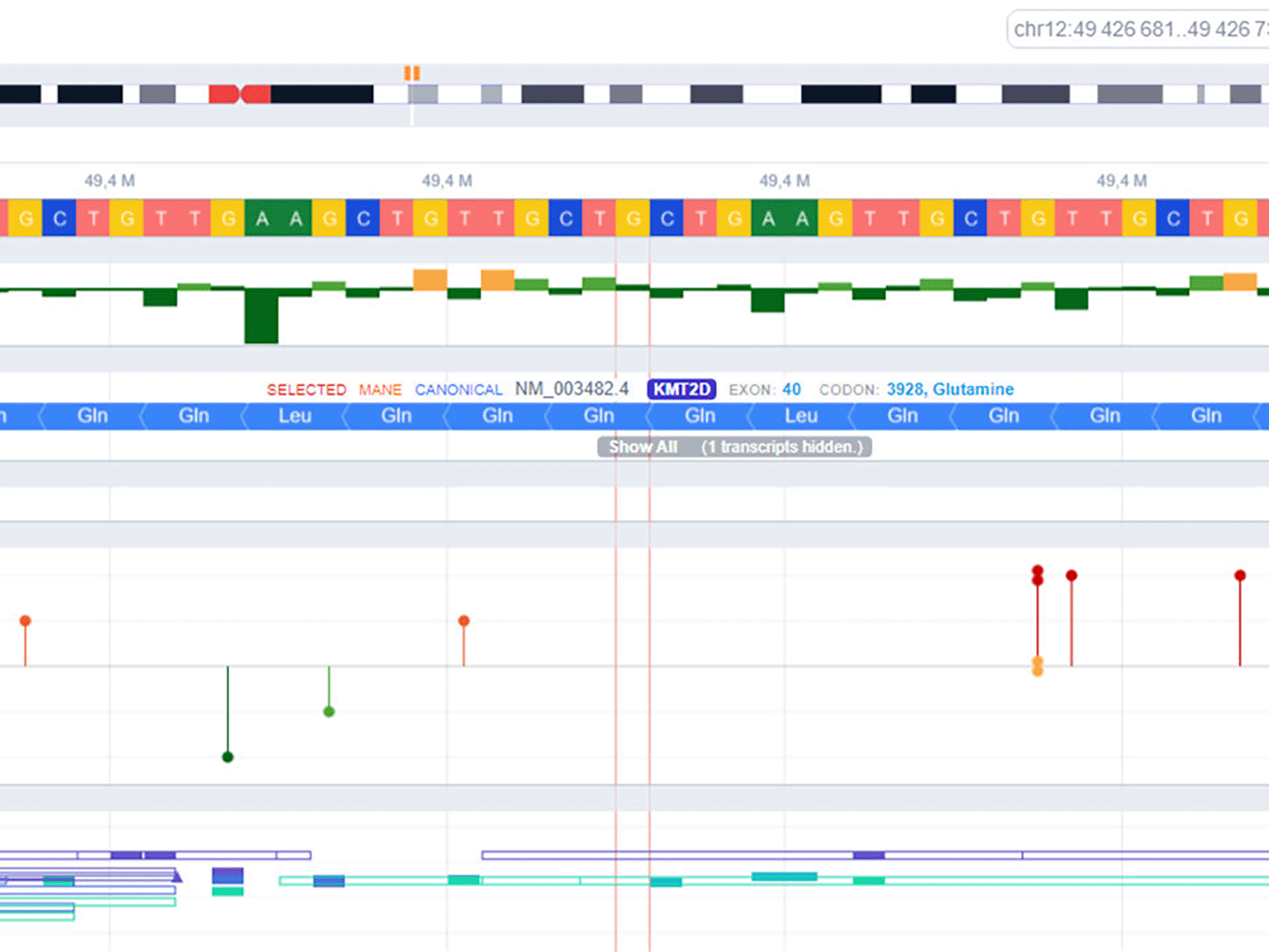
As part of the genetic investigation, a postnatal karyotype was performed, resulting in 46, XY normal formula. To confirm the diagnosis, DNA extraction was carried out and high-throughput sequencing using the MiSeq sequencer was performed, thanks to a custom panel containing genes implicated in Kabuki syndrome as described in the literature. Subsequently, bioinformatic analysis was completed for variant annotation, filtration, and classification. A variant in the KMT2D gene (NM_003482.4): c.11782C>T (p.Gln3928Ter) was identified, representing a likely pathogenic class 4 missense point mutation according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) (Figure 7).
Kabuki syndrome presents a high interindividual clinical variability.3 The initial clinical presentation varies from person to person.4 Growth retardation is almost always present. In our patient, stature growth retardation was observed from the age of six months, but there was no weight retardation. Auditory abnormalities are observed in 20 to 30% of cases5 but were not observed in our patient. These abnormalities can be due to conductive, sensorineural, or mixed hearing loss. Ophthalmological abnormalities such as strabismus, ptosis, and corneal anomalies can impact vision.5 In our study, our patient presented with convergent strabismus and myopia. Hypotonia is an important sign in Kabuki syndrome, often noted from birth and persisting for several years,6 often associated with swallowing difficulties due to hypotonia. In our patient, hypotonia and swallowing difficulties were absent. The most common musculoskeletal abnormalities in Kabuki syndrome are brachydactyly, brachymesophalangy, clinodactyly of the 5th finger, scoliosis, hypermobility, or dislocation of certain joints.7 Musculoskeletal abnormalities were mostly consistent with the literature, except for scoliosis, which was not present in our patient.
Cardiac involvement is found in 50% of patients.8 The most common malformations are atrial or ventricular septal defects and aortic coarctation. In our observation, a restrictive ventricular septal defect was noted, but without cardiac impairment. Renal abnormalities have been observed in 25% of Kabuki syndrome patients.8 These abnormalities include hydronephrosis, abnormal kidney position, and renal dysplasia. In our observation, the patient had malformative uropathy in the form of pyeloureteral junction syndrome without involvement of the upper urinary tract. In the literature, the association of pyeloureteral junction syndrome with Kabuki syndrome has not been reported. Growth hormone (GH) deficiency or hyperinsulinism have been described in 15 to 25% of cases.4,7 In our case, the patient suffered from GH deficiency and was successfully treated with hormone replacement therapy. Gastrointestinal and hepatic abnormalities are described in only 5% of Kabuki syndrome patients. These can include intestinal malrotation, anorectal anomalies, ano-vestibular fistulas, biliary atresia, and hepatic fibrosis.6 In our case, no gastrointestinal or hepatic abnormalities were noted. Sixty percent of patients develop infections, mainly chronic otitis media, upper respiratory tract infections, and pneumonia. This increased susceptibility to infections could be due to anatomical or immunological abnormalities.8 There is an associated immune deficiency with IgA deficiency and hypogammaglobulinemia.9 Our patient suffered from recurrent acute bronchiolitis before the age of two, with signs of maxillary sinusitis on brain CT scan. An immune assessment was performed on our patient and showed no abnormalities.
The diagnosis of Kabuki syndrome is usually suspected based on clinical presentation but confirmed through genetic testing. The majority of affected patients are sporadic cases, but there are some cases in the literature with autosomal dominant transmission.9,10 The KMT2D or MLL2 gene, also known as ALR (ALL1-related gene), is the most well-known gene responsible for Kabuki syndrome. It is located on chromosome 12. The encoded protein is a histone-3-lysine-4-methyltransferase. The clinical heterogeneity found in this disease can be explained by the pleiotropic effect of KMT2D and its variable expression.11,12 A 2010 exome sequencing study of 10 patients with Kabuki syndrome identified the KMT2D gene as a major cause.13 According to several studies, a mutation in the KMT2D gene is found in 56 to 75% of cases,13,14 thus determining Kabuki syndrome type 1.9 Nonsense mutations are the most frequent. Furthermore, there are rare cases of mosaic mutations that are identified during Kabuki syndrome.15 The second gene responsible for Kabuki syndrome is KDM6A (or UTX), located on the X chromosome, that codes for the KDM6A protein, responsible for Kabuki syndrome type 2.9 There is an interaction between the two proteins KDM6A and KMT2D.16 Facial dysmorphia is more pronounced in type 1, while cardiovascular abnormalities are more frequent in type 2. The diagnostic approach in cases of Kabuki syndrome requires initial screening for mutations in the KMT2D gene since it is the most frequent. If the clinical presentation strongly suggests it, further screening for mutations in the KDM6A gene or large DNA rearrangements may be necessary if no mutations are found in the KMT2D gene. In our case, we identified the KMT2D variant (NM_003482.4): c.11782C>T (p.Gln3928Ter), which is a point mutation by substitution in the KMT2D gene, a likely pathogenic nonsense mutation of class 4 according to the recommendations of the American College of Medical Genetics and Genomics “ACMG,” and not reported in the “Clinvar” database (Figure 7). Finally, Sanger sequencing was performed to confirm the mutation. This genetic mutation determined a phenotype characterized by the absence of neonatal hypotonia or swallowing disorders,9 the absence of frequent immune deficiency seen in other phenotypes, the absence of digestive abnormalities, the presence of facial dysmorphia typical of type 1 associated with a more frequent cardiac malformation in type 2,9 the presence of an endocrinopathy such as GH deficiency, which responds to usual doses of replacement hormones unlike other genetic abnormalities such as Turner syndrome,17 and an average intellectual level as described in the literature.17 Furthermore, there are several differential diagnoses that can delay the positive diagnosis, such as CHARGE syndrome characterized by a cleft palate, which may be present in Kabuki syndrome but absent in our case.18 Another possibility is 22q11 deletion syndrome characterized by urinary tract malformations but presenting different facial features.18 There is also Crouzon syndrome characterized by ventriculomegaly, similar to our case, but with facial dysmorphia characterized by a prominent forehead and hypoplastic nasal bone. Therefore, genetic testing is essential for a definitive diagnosis.19
Finally, this case report’s strength resides in the coherence of the diagnostic process and the long-term surveillance enabling us to describe all the clinical abnormalities’ evolution in time.
The diagnosis of Kabuki syndrome is clinical. However, confirmation through genetic testing is necessary to optimize patient management, establish a rapid screening strategy, and especially to address the multiple complications associated with the syndrome. Kabuki syndrome is characterized by phenotypic heterogeneity due to the various mutations described. The syndrome is inherited in an autosomal dominant manner, highlighting the importance of genetic counseling. Prenatal diagnosis is increasingly recommended, even though the risk of recurrence of this condition is low.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)