Introduction
What pushed you over the edge, huh? I bet it started with something small: a little too much to drink, sitting in traffic getting a lung-full of diesel exhaust or a drafty window in your apartment. Maybe your boss starts making fun of you like that bully did when you were in middle school. Whatever the insult, it’s not a big deal if it only happens once. You shake it off. But it just … keeps … happening. Once you could take. But this? No. You broke, and you broke bad. And now we’re here in this grimy room that smells like cheap, week-old coffee and donuts made from dirty gym socks. Neither of us wants to be here, and we don’t have to be for long. I just want to know two, simple things: why are we here and how did you do it?
Well, this isn’t simple. Not all criminals are the same. Some are serial killers or arsonists, while others are burglars or blackmailers. It’s not just people that break bad; fibroblasts do it, too, and in just as many ways. They can be like Avon Barksdale, Marlo Stansfield or Bodie enabling neutrophils, macrophages and other innate immune cells to be the Bubbles of fibrotic diseases (FD). Or they can be like Viktor Bout and Yevgeny Prigozhin, organizing and arming forces of T, B and dendritic cells (DCs) in immune-mediated inflammatory diseases (IMID). Arson isn’t too different from how fibroblasts destroy articular cartilage in rheumatoid arthritis (RA). Poe’s Montresor immured Fortunato which, in a way, is what fibroblasts do in FD. As many ways in which people commit crimes, so too can fibroblasts be pathogenic in FD and IMID. In this review we will largely focus on IMID and sterile FD not associated with tumors. Although fibroblasts interact with many types of cells, this review will largely cover their relationships with leukocytes and the extracellular matrix (ECM).1–13 By the end of this, you’ll know some of the reasons why fibroblasts break bad, what drives them do it and a few of the questions we still need to answer.
Recruitment and positioning of specialized leukocytes
Leukocyte infiltrates are common across diseases that clinically manifest as either FD (e.g. idiopathic pulmonary fibrosis (IPF), non-alcoholic steatohepatitis (NASH) and systemic sclerosis (SSc)) or IMID (e.g. rheumatoid arthritis (RA), Sjogren’s syndrome (SS) and discoid lupus erythematosus (DLE)). However, the magnitude, composition, density and organization of leukocyte infiltrates differ. Generally, the number of infiltrating leukocytes per mm3 appears to be lower in FD than IMID biopsies. Infiltrates in FD are generally dominated by innate immune cells like neutrophils and macrophages with smaller proportions of innate and tissue resident lymphocytes. IMID generally feature lymphocyte-rich infiltrates and aggregates. However, relatively few studies have directly compared the frequencies of lymphocyte aggregates between FD and IMID, and there is considerable histological heterogeneity.14–22 This comparison is further complicated in FD by changes in the composition and size of infiltrates as tissues become more fibrotic.23
Regardless of the insult or injury that initiates FD or IMID, the earliest events are likely shared between these diseases. These likely include the release of alarmins, damage associated molecular patterns (DAMPs) and a local increase in vascular permeability that results in a serum response ((Figure 1A) reviewed in Refs. 24–26). Endothelial and myeloid cells express a wide repertoire of pattern-recognition receptors (PRRs) that recognize DAMPs, but fibroblast PRR expression appears to be more tissue-specific (reviewed in Ref. 27).3,28–32 Fibroblasts are more widely responsive to alarmins and serum.33–48 Although tissue-specific fibroblast responses to PRR signaling likely contribute to the sites and outcomes of FD and IMID, we focus on the more universal alarmin response here. Among the alarmins that fibroblasts respond to are IL-1α, IL-1β, and IL-33.
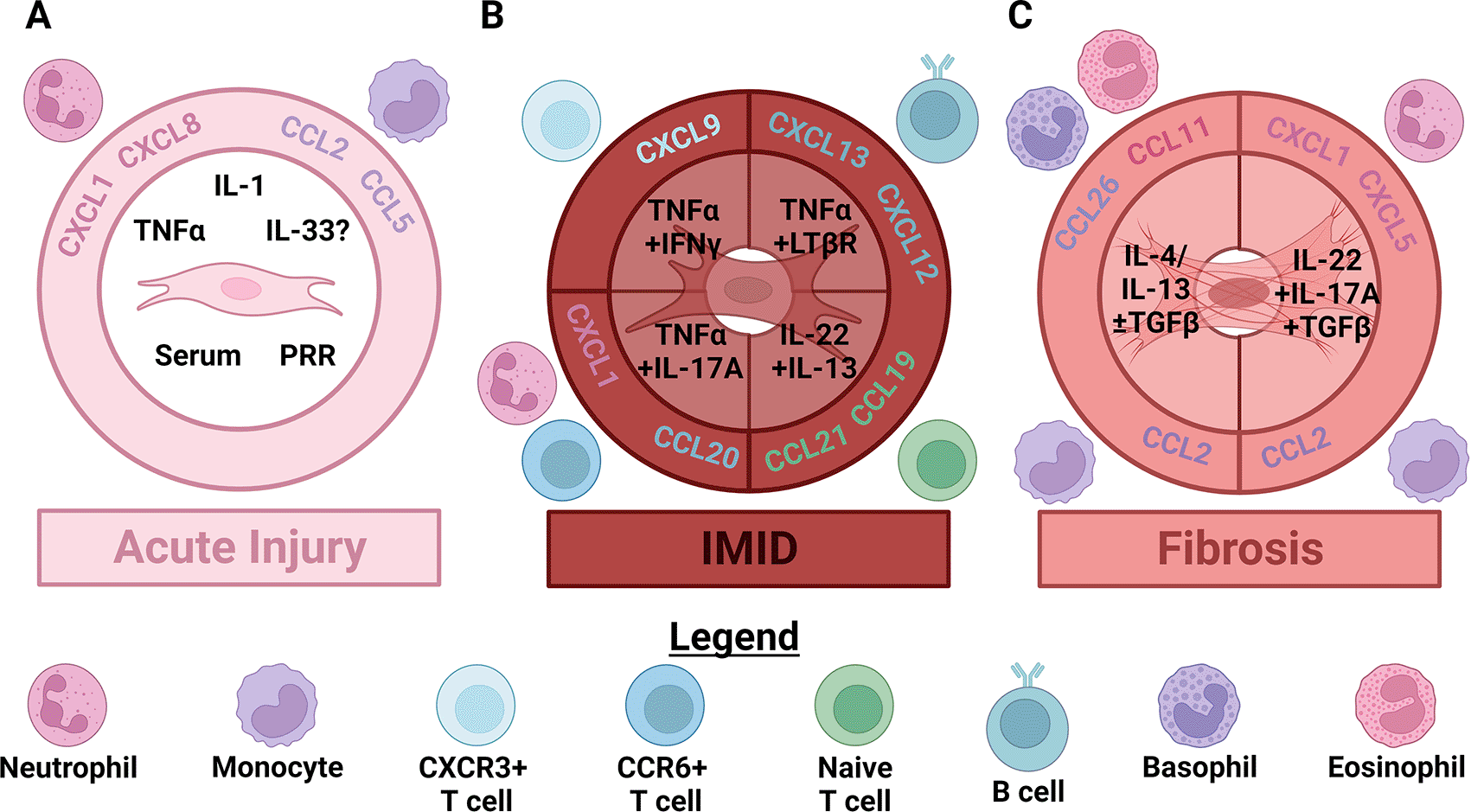
Figure 1. As acute injury progresses to chronic disease fibroblasts orchestrate changes in the size and composition of leukocyte infiltrates by producing distinct chemokines.
(A) In acute injury, damage signals like serum, pattern recognition receptor (PRR) activation, interleukin (IL)-1 and tumor necrosis factor α (TNFα) activate fibroblasts. Whether fibroblasts are directly activated by IL-33 is still controversial. Fibroblasts activated by these signals produce chemokines that attract neutrophils like CXCL1 and CXCL8, as well as chemokines that attract classical monocytes like CCL2 and CCL5. (B) In immune-mediated inflammatory diseases (IMID) activated innate lymphocytes (including innate lymphoid, γδ T, NK, iNKT and MAIT cells) begin producing polarizing cytokines. This initiates local production of specialized chemokines which induce extravasation of the first polarized, memory αβ T cells that increase polarized cytokine production. In type 1 skewed responses this first wave of lymphocytes produces TNFα and IFNγ, which synergistically stimulate fibroblast secretion of CXCL9, CXCL10 and CXCL11. These cytokines attract more CXCR3+ type 1 or type 3 skewed T cells. IFNγ also appears to inhibit TNFα-induced production of neutrophil chemokines by fibroblasts. Fibroblasts instead stimulated by type 3 cytokines including TNFα and IL-17A continue secreting neutrophil chemoattractants like CXCL1 and begin producing additional cytokines like CCL20. These cytokines attract additional CCR6+ type 3 skewed T cells. Accumulation of enough activated dendritic cells and T cells leads to significant TNFα/TNF receptor 2 and lymphotoxin α1β2/lymphotoxin β receptor (LTβR) signaling. Activation of these pathways in fibroblasts can induce an additional stage of specialization that recruits naïve αβ T cells via secretion of CCL19 and CCL21, and B cells through CXCL13 and/or CXCL12. (C) Relatively few αβ T cells accumulate in sterile fibrotic diseases (compared to IMID) leading to distinct signaling and fibroblast differentiation. In fibrotic diseases largely characterized by type 2 skewing, IL-4 and/or IL-13, especially in concert with active TGFβ, induce secretion of CCL11, CCL26 and CCL2 by fibroblasts. CCL11 attracts eosinophils, CCL26 attracts basophils and CCL2 continues to attract classical monocytes. Conversely, in fibrotic diseases that exhibit more type 3 skewing IL-22 and IL-17A, in combination with TGFβ stimulate secretion of neutrophil chemokines, like CXCL1 and CXCL5, and CCL2 by fibroblasts. Note that most human IMID and fibrotic diseases are characterized by mixed, rather than purely polarized, inflammation. Figure created with BioRender.
Measuring alarmins in patients is complex due to the lack of early diagnostics, heterogeneity in disease severity and course, medications, genetic background and other factors. Despite these challenges, increased expression of alarmins is associated with both FD and IMID although data from patient samples is relatively scarce. Increased IL-1β, IL-33 and TSLP protein was detected in the bronchoalveolar lavage fluid (BALF) of patients with IPF and SSc, although there was considerable variability between cohorts.49–53 Higher levels of IL-1α were also observed in the serum from some SSc patients.54 SSc patients treated with bermekimab, an IL-1α antagonist antibody, in a 12-week phase 2A trial had better lung function and fewer swollen joints supporting a pathogenic role for this cytokine in some patients.55 Synovial fluid from RA patients had higher levels of IL-1β than osteoarthritis patients, although the detected levels were lower than TNF and IL-6.56–58 This difference in local expression may partly explain the modest ACR score improvements in RA clinical trials of Anakinra compared to TNFα antagonists and tocilizumab.59,60 Higher serum levels of IL-33 were observed in a subset of RA patients, which was associated with pulmonary complications and more severe disease.61,62 Elevated synovial fluid IL-33 also distinguished RA patients from those with osteoarthritis.62,63 These data suggest that alarmins can activate fibroblasts in either FD or IMID and may be pathogenic, although the biomedical community would benefit from higher-powered studies in genetically diverse patient cohorts.
IL-1α is expressed by various cell types in the cytoplasm and nucleus and can be released during cellular damage without any modification. In contrast, immature IL-1β must be processed by a specialized complex known as the inflammasome. The inflammasome is a supra molecular organizing complex comprised of adapter proteins and active caspases. Formation of the complex results in caspase mediated cleavage of IL-1β and the gasdermins that form the membrane pores required for the release of mature IL-1β (reviewed in Ref. 26). Both IL-1α and IL-1β bind to IL-1R1 on fibroblasts to elicit secretion of a variety of chemokines. Stimulation of fibroblasts with recombinant IL-1α in vitro induced CXCL8 RNA within 30 minutes and secretion of bioactive CXCL8 and CCL2 within 24 hours (Figure 1A). Conditioned media from damaged lung epithelial cells induced transcription of these same chemokines, which required IL-1α, not IL-1β. In this case, IL-1α-driven transcription of these chemokines synergized with TLR3 signaling. This required at least JAK2, TAK1 and IKK2.36,44,45 IL-1β induced production of CXCL8 and CCL2 as well as CXCL1 and CCL5/RANTES (Figure 1A).46,48 Together, these data show that fibroblasts from multiple sites can respond to the alarmins IL-1α and IL-1β by secreting neutrophil and monocyte chemokines.
Do these in vitro findings translate in vivo? Alum injection demonstrated that IL-1α-/- mice showed a partial decrease in neutrophil infiltration into the site of injection, while IL-1α-/- IL-1β-/- mice showed complete ablation of neutrophil infiltration indicating some redundancy between these cytokines.64 Murine myocardial infarction results in large monocyte and neutrophil infiltrates into the damaged heart, which required IL-1 signaling in non-myeloid cells.65 Conditional deletion of IL-1R1 using the Col1a2-CreER(T) line in this model reduced cardiac leukocyte infiltration, although the authors did not specifically quantify neutrophil or monocyte infiltrates.47 Acute, diphtheria toxin-mediated depletion of Kupffer cells similarly demonstrated that IL-1α, IL-1β and TNFα activate hepatic stellate cells (fibroblastic cells found in the liver) and liver sinusoidal endothelial cells. Both cell types rapidly produced Ccl2 to recruit monocytes and replenish the Kupffer cell niche.66 Together these results suggest that IL-1α and IL-1β can stimulate fibroblast-mediated recruitment of neutrophils and monocytes in vivo. These data suggest that chemokine production by fibroblasts is important for monocyte and granulocyte recruitment in response to IL-1. However, a direct demonstration would require conditional, fibroblast-specific IL-1R1 knockout and quantification of monocyte and granulocyte infiltration.
Less is known about fibroblast responses to IL-33 than IL-1α and IL-1β. Human and murine fibroblasts can express IL-33 in vivo and in vitro.62,67–76 As a nuclear and secreted factor that undergoes proteolytic processing, IL-33 biology is complex and requires further experimental clarification (reviewed in Ref. 77). The current picture is that homeostatic murine adventitial fibroblasts in several organs and differentiated lymphoid organ fibroblasts express IL-33 in vivo.67–69,71,72 Either this is different for human fibroblasts or IL-33 expression decreases with traditional fibroblast culture methods: human fibroblasts required stimulation to induce IL-33 expression in vitro. Combined stimulation by TNFα and IFNγ triggered IL-33 expression by dermal fibroblasts, which was further enhanced by TLR3 activation.76 Either TNFα or IL-1β alone stimulated IL-33 production by synovial fibroblasts from RA patients, which also synergized with TLR3 activation.74,75 However, secreted IL-33 was rarely detected in these experiments.73–76 Does this mean that fibroblasts express nuclear IL-33 but cannot secrete it?
It is possible that, under the conditions used in these studies, the cultured fibroblasts did not secrete IL-33. However, because conditional deletion of IL-33 from fibroblasts and ST2 from leukocytes in vivo yielded similar phenotypes, it seems likely that fibroblasts can secrete IL-33.78,79 There are two alternative explanations that are also plausible. First, oxidization of IL-33 after secretion prevents binding to its receptor IL1RL1/ST2 and subsequent signaling. Distinct antibodies are required to detect oxidized IL-33 versus reduced IL-33.80 Thus, the lack of detectable secreted IL-33 in these studies may have been due to this conformational change. This, however, seems less likely than the second alternative explanation. Alternative splicing of IL1RL1 yields both transmembrane and soluble proteins. The soluble form antagonizes IL-33 signaling.81 Proteolytically processed IL-33 is a small protein (~175-161 amino acids).82 Although not conclusively demonstrated for most IL-33 antibodies, it seems likely that the binding of IL-33 to soluble IL1RL1 would cross-block subsequent detection by anti-IL-33 detection agents. Production of soluble IL1RL1 by fibroblasts would thus impair detection of secreted IL-33 from these cells. Further empirical dissection of IL-33 secretion by fibroblasts would provide useful clarification.
Regardless of whether fibroblasts secrete IL-33 they can express the protein. Since the most robust evidence is for intracellular expression of IL-33, does this form exert effects on fibroblasts? When expressed in the nucleus, full-length IL-33 appears to repress transcriptional activation by NF-κB. This inhibition translated to reduced secretion of CXCL8 and CCL2 by RA synovial fibroblasts stimulated with TNFα alone or the combination of TNFα, IL-1β and a TLR3 agonist.74,75 Thus, induction of intracellular IL-33 expression by IL-1 and TNFα may act as a negative feedback regulator of acute inflammatory responses by fibroblasts. Many cells aside from fibroblasts can secrete processed, highly active IL-33. Is there evidence that fibroblasts are impacted by this secreted form?
Data from RA synovial fibroblasts suggests that exogenous IL-33 may have the opposite effect from that of intracellular IL-33. Stimulation with recombinant IL-33 increased secretion of CXCL8 and transcription of several other genes.73 Blocking IL1RL1 inhibited induction of some of these by IL-33 suggesting that fibroblasts may be activated by IL-33.74 However, IL-33 also increased transcription of IL-1β in RA synovial fibroblasts.73 As IL-1β can stimulate many of these same transcripts it is currently unclear which effects were the direct result of IL-33 signaling and which were indirectly stimulated by IL-33-driven IL-1β production (Figure 1A). This is somewhat controversial as another study was unable to detect direct effects of exogenous IL-33 on the RA synovial fibroblasts they used.75 All in all, it seems likely that fibroblasts can respond to IL-33 under some circumstances, however the effects and relative importance of this require further work.
Correlating with the elevated alarmin expression in patients with FD and IMID, levels of the alarmin-stimulated chemokines CCL2, CCL5, CXCL2 and CXCL8 were also higher patients with FD and IMID than healthy controls.83–88 In addition to producing these chemokines in response to IL-1α and IL-1β, fibroblasts can produce these when stimulated with TNFα (Figure 1A). This is true for human cells and animal models of both FD and IMID and occurs both in vitro and in vivo.17,22,85,89,90 And in patients with inflammatory bowel disease (IBD) a fibroblast IL-1 response signature is correlated with local neutrophil infiltrates and resistance to both TNF antagonists and steroids.37 This supports the redundance of IL-1 and TNFα in a subset of patients. These patients would likely benefit from combination therapy targeting these redundant signaling systems. If alarmins and DAMPs are produced in both FD and IMID, why are the infiltrate compositions generally so different between theses distinct clinical disease manifestations? The answers to these questions are currently unclear, although they likely involve the results of combinatorial signaling events, which will be explored next.
During the priming phase of antigen-specific responses, including IMID, immature DCs and tissue-resident macrophages are also activated by alarmins and PRR. Activated perivascular macrophages secrete chemokines like CXCL2 that initially attract maturing DCs towards blood vessels. Without an influx of activated antigen-specific T cells (or the presence of antigen-specific tissue resident T cells), these maturing DCs leave the damaged tissue through the draining lymphatics and prime antigen-specific naïve T cells in lymph nodes.91–93 Similarly, in sterile FD where tolerance appears to be maintained any DCs that are activated during the initial insult likely migrate to the draining lymph nodes but fail to induce a T cell response.94,95 How these discrepant responses occur is an interesting question, but beyond the scope of this review. Simultaneously, neutrophils and monocytes are drawn into the tissue by the chemokines produced in response to alarmins, PRR signaling and/or the serum response as described previously.
At this early stage, the monocytes likely differentiate into a mixture of macrophages and DCs. In IMID, the activated T cells proliferate, differentiate, exit the lymph node and enter the damaged tissue through activated post-capillary venules.96,97 These T cells are restimulated by perivascular DCs presenting cognate antigens, and these reactivated T cells retain the DCs in the affected perivascular or peri-damaged space.91,98–103 Retention of these DCs and the initial accumulation of activated antigen-specific T cells likely leads to nucleation of lymphocyte aggregates and infiltrates where lymphocytes predominate. Conversely, the lack of antigen-specific T cell influx and DC retention in FD leads to an entirely different set of signals. This, we propose, is where fibroblast activation states and functions in FD and IMID diverge.
Within hours of damage or insult fibroblasts in non-lymphoid tissues turn off their homeostatic programs and begin expressing Podoplanin (PDPN).104,105 PDPN expression by human fibroblasts from multiple sites is induced by acute inflammatory stimuli like IL-1β and TNFα, consistent with this activation occurring prior to reparative or fibrotic signals like TGFβ.104,106,107 Unliganded PDPN initiates fibroblast contraction by binding to ezrin, activation of Rho kinases (ROCK) and phosphorylation of ezrin/radixin/moesin family proteins and myosin light chain.108–110 Fibroblast contraction can activate latent TGFβ through integrin αV heterodimers. Active TGFβ is both a suppressor of generally anti-fibrotic type 1 immune skewing and one of the most potent pro-fibrotic signals.111–116 Therapeutic inhibition of integrin mediated latent TGFβ activation in fibrotic diseases is being pursued by multiple companies including Pliant Therapeutics.117 Although TGFβ suppresses signals associated with type 1 immune polarization it appears to spare production of myeloid and neutrophil chemokines by fibroblasts, at least as part of the serum response and in combination with TLR4 or IL-17A.118 Contraction also increases the local stiffness of the ECM which can initiate durotaxis and tension responses through integrins.119
Durotactic attraction between myofibroblasts and macrophages is believed to be more effective than chemotaxis and haptotaxis.119,120 Macrophages are especially important collaborators in generating the fibrotic niche: genetic or chemical depletion of macrophages abrogates fibrosis progression.121–128 Once in proximity fibroblasts and macrophages establish strong adhesion through homotypic Cadherin-11 (CDH-11) contacts. This creates a pro-fibrotic niche where latent TGFβ produced by macrophages can be activated by myofibroblast contraction.129 Thus, therapies aimed at limiting mechano-sensing in fibroblasts and macrophages are currently being explored. These include inhibition of PDPN, ROCK1/2 and focal-adhesion kinase (FAK) as well as strategies to dissociate fibroblasts from macrophages (e.g. CDH-11 antagonists). Immune cells other than macrophages can also sense mechanical forces, which similarly impact their activation and function.130,131 Therefore, it is possible that neutrophils, eosinophils and others are recruited to the pro-fibrotic niche through the same mechanisms. Similarly, there are mechanisms other than PDPN that activate fibroblast contractility which likely play important roles in the fibrotic activation of fibroblasts.132–135 Thus, the combination of contraction-induced durotaxis and selective sparing of myeloid and granulocyte chemokine expression already appears to differentiate fibroblast functions in FD from those in IMID.
Unlike the macrophages and neutrophils that predominate in fibrosis-associated leukocyte infiltrates, DCs express the PDPN ligand CLEC-2, which decouples PDPN from ezrin and inhibits fibroblast contraction.108,109,136 This is one way that early differences in the composition of leukocyte infiltrates may result in different fibroblast functions and clinical pathologies in IMID and FD. Whether this is true in vivo requires experimental validation, though, as CD177, which is expressed by neutrophils, binds to PDPN and may exert similar effects.110 Relaxation of fibroblasts around injured areas through interaction with DCs is likely necessary to accommodate early infiltration of T cells like the swelling and expansion of lymph nodes during immune responses.9,10,108,109,137–139 In addition, these DCs may provide TNFR2 or LTβR ligands to trigger further fibroblast specialization.140–145 Some sites, like the lung, seem to require local retention of DCs for sufficient secondary fibroblast activation and lymphocyte accumulation regardless of whether the response is type 1 or type 3 skewed.146–152 In others, like the salivary glands, IL-22 and IL-13 produced by γδ and iNKT cells and ILC2s may be sufficient to initiate lymphocyte accumulation and secondary fibroblast activation. Sustained lymphocyte recruitment in this case appears to require an influx of IL-22 producing αβ T cells.107,153,154 Regardless of the site and activating signals, this second round of fibroblast specialization in IMID seems to involve fibroblast proliferation and the induction of T cell recruiting chemokines by these cells.105,107,146–159
Patterns of lymphocyte infiltrates can vary from diffusely spread throughout the tissue to tightly aggregated.14,160–163 Aggregates frequently form around microanatomic structures such as blood vessels and pulmonary airways.14,67,163,164 Among diseases with T cell aggregates, their size and organization can vary between small (~5-20), medium (~20-50) and large (>50).14,162 Small T cell aggregates can likely be recruited by activated perivascular DCs without the help of specialized fibroblasts, but this may differ between mice and humans.14,91,98 Growth of T cell aggregates beyond ~10 cells appears to require the expansion of specialized fibroblast networks.14,91,156–158,164 As polarized, activated memory T cells enter an inflamed tissue, or as ILCs and other tissue-resident lymphocytes are activated, the combination of innate and adaptive cytokines likely tunes the fibroblast chemokine program to attract the appropriate lymphocytes for the response.
In the case of type 1 polarization, the synergistic combination of TNFα or IL-1 and IFNγ suppresses neutrophil chemokines in vitro while increasing production of CXCR3 ligands via cooperation between STAT1 and Nuclear factor-κB (NF-κB, Figure 1B).165–169 Fibroblasts expressing CXCL9 are important for CXCR3+ TH1 differentiation in lymph nodes, and similarly activated fibroblasts expressing multiple CXCR3 ligands are increased in the salivary glands of SS patients, synovia of RA patients and in tumors.70,170–174 Likely these activated fibroblasts contribute to extravasation of type 1 or type 3 lymphocytes expressing CXCR3 (Figure 1B).98,175–178 Conversely, combined stimulation of fibroblasts with the type 3 stimuli TNFα and IL-17A decreased CCL3 and CCL5 secretion while increasing expression of CCL20 and neutrophil chemoattractants.90,179–181 Whether a similar fibroblast state occurs in patients awaits experimental verification. These fibroblasts are likely adapted to draw in type 3 polarized CCR6+ lymphocytes and neutrophils (Figure 1B).178,182 Specialized adventitial fibroblasts can indirectly instigate type 2 lymphocyte recruitment by creating specialized niches for ILC2s and eosinophils.67 These observations suggest an evolving dialogue between leukocytes and fibroblasts that can recruit specialized lymphocytes in IMID with distinct immune polarization. The relative importance of fibroblast production of these chemokines requires experimental investigation in vivo.
The recruitment of polarized lymphocytes by fibroblasts appears to occur early in the growth of aggregates. Small lymphocyte aggregates are dominated by CD45RO+ T cells, which have likely been polarized during their activation in secondary lymphoid organs (SLO). Further fibroblast specialization seems to be required to recruit B cells and naïve T cells as the frequency of B cells and naïve T cells increases with aggregate size.14,156,157,164 Extravasation and localization of naïve T cells into SLO requires secretion of the CCR7 ligands CCL19 and CCL21 by fibroblasts.70,183–187 Similar fibroblasts expressing CCL19 and CCL21 emerge in several IMID and tumors, and also appear to correlate with T cell infiltration and lymphoid aggregate size.21,156–158,171,188–192 In patients with interstitial pneumonia with autoimmune features, BALF CCL19 concentrations and lymphocyte numbers were positively correlated supporting CCL19-induced pulmonary lymphocyte infiltration. Longitudinal tracking of these patients demonstrated that high baseline BALF CCL19 and lymphocytes were associated with a lack of pulmonary fibrosis up to one year later. This observation supports divergent fibroblast activation occurring in IMID versus FD, and that the signals that induce these states may cross-inhibit each other.193 What induces fibroblast production of CCL19 and CCL21 in IMID is also unclear, but the clues we have are discussed below.
In lymphoid organs, there is evidence for TNFR1/2 and LTβR ligands driving expression of these chemokines (Figure 1B). Pharmacological or genetic antagonism of LTβR signaling reduces or prevents CCL21 and CCL19 expression in models of IMID.107,140,141,145,184,192,194–197 However, expression of CCL21 in the lungs appears to be independent of both TNFR1/2 and LTβR signaling.198 We are unaware of in vitro confirmation of CCL19 and CCL21 induction in fibroblasts by these signals. Larger reductions in CCL19 RNA were seen in RA patients with EULAR responses to disease-modifying anti-rheumatic therapies (DMARDs) than those who failed to respond, which suggests a potential association with pathogenesis.199 But whether fibroblasts drive significant disease pathology by producing these chemokines remains an open question.
B cell infiltrates into IMID tissues are generally less common than T cell infiltrates.14,156,188 However, in some IMID like SS, RA and DLE, large B cell infiltrates occur in substantial proportions of patients (approximately 42% in RA, 89% in SS and 92% in DLE) although some of these cohorts were relatively small.14,156,157 Aggregates containing both T and B cells can either be non-segregated, with T and B cells evenly distributed throughout, or segregated, with discrete T cell zones and B cell follicles similar to SLO.14,21,156,157,164,192,200,201 In this review we define “tertiary lymphoid structures” (TLSs) as the subset of lymphocyte aggregates with these discrete T cell zones and B cell follicles. Infiltration of B cells in IMID, especially smaller infiltrates that lack T-B cell separation, occur in the absence of (or preceding) mature follicular dendritic cells (FDC, B cell supporting fibroblasts in SLO).14,156,157 Regardless of FDC maturation status, CXCL13 generally appears necessary for inflammation-associated increases of B cells in peripheral tissues (Figure 1B).156,157,202–206 However, in specific cases like the lung B cell infiltrates can occur in the absence of CXCL13 and appear to require CXCL12 (Figure 1B).150 This could suggest that CXCL12 is sufficient for B cell extravasation and localization, or that CXCL13 negative pulmonary TLSs are exclusively populated by resident memory B cells.207,208
In IMID where CXCL13 is induced, expression by myeloid and peripheral helper T cells (TPH) is likely sufficient to drive the initial accumulation of a few memory B cells.156,157,202,203,209 As inflammation progresses, however, expression of CXCL13 by fibroblasts appears important for generating local germinal centers.14,156,157,162,203,210 During SLO development, CXCL13 expression can be induced by or independent of TNFR1/2 and LTβR signaling depending on the site and timing (Figure 1B).145,195,211–213 Similarly mixed observations have been made in murine models of IMID and lymphoid aggregate formation. Antagonizing LTβR signaling was sufficient to reduce or inhibit CXCL13 production in the non-obese diabetic (NOD) model of SS and a model of autoimmune pancreatitis.192,194,197 IL-22 production is necessary for induction of CXC13 by salivary gland and lung fibroblasts, but the salivary gland induction depends on LTβR while the lung does not.154,214 Together, these data suggest that a stepwise dialogue between leukocyte infiltrates and fibroblasts causes fibroblast differentiation that may separate IMID from FD.
Alarmins induced neutrophil and monocyte chemokine secretion by fibroblasts as well as contraction-induced durotaxis. This first fibroblast activation state is likely similar between IMID and FD (Figure 1A). Next, in IMID a first wave of activated, polarized T cells infiltrates the tissue resulting in local retention of DCs and fibroblast relaxation. Collaboration between TNFR and LTβR ligands produced by DCs with adaptive cytokines produced by T cells, ILCs, NK, MAIT and iNKT cells results in a second step of fibroblast differentiation. Second-stage IMID fibroblasts likely begin to express CCL19, CCL21, CXCL12 and CXCL13, which recruits B cells and naïve T cells (Figure 1B). Perhaps, as recently described, ligation of ICOSL on fibroblasts or CD40/CD40L-dependent TPH/B cell interactions are necessary for further lymphoid aggregate expansion and compartmentalization.196,215 Conversely, in FD the monocyte- and neutrophil-rich infiltrates promote fibroblast contractility and stiffening of the matrix. This leads to the activation of latent TGFβ, which combines with alarmin signaling to continue monocyte and neutrophil extravasation while preventing T cell infiltration (Figure 1C). From these observations, divergent paths of fibroblast activation between FD and IMID begin to emerge.
Rationing of growth factors
From the previous section it appears that fibroblasts can contribute to differential leukocyte extravasation in FD and IMID. However, the number of cells that can occupy a tissue is regulated by the availability of growth and survival factors.216–219 This observation raises our next two questions. First, having contributed to leukocyte infiltration, do fibroblasts provide survival signals to these cells in FD and IMID? The answer, as this section will demonstrate, appears to be yes. These survival signals can take the form of soluble factors, transmembrane molecules or modifications of the ECM. We will concentrate on secreted growth factors in this review. Second, do fibroblasts sense and regulate cell composition by changing the types and quantities of growth factors they provide? This is an active area of research, but initial data suggests that they do.
In line with the correlation between elevated alarmin levels and monocyte and granulocyte chemokines in FD and IMID, growth factors to support these innate leukocytes are also elevated in some diseases. GM-CSF but not G-CSF was higher in synovial fluid of RA versus OA patients, and there appeared to be an inverse relationship between synovial fluid GM- and G-CSF concentrations.220–222 Levels of GM-CSF correlated with IL-1β and TNFα quantities in the synovial fluid of RA patients, and both IL-1β and TNFα induced secretion of GM-CSF by synovial fibroblasts.222 GM-CSF protein levels were higher in urine from patients with active lupus nephritis compared to inactive systemic lupus erythematosus, other chronic kidney diseases and healthy controls.223 G-CSF was detectable in tears from both IgG4-related disease and SS patients, however levels in SS patients did not appear to differ from healthy controls.224–226 Data on growth factor levels in local fluid and biopsy samples in FD is scarcer than for IMID. What is available shows a trend towards increased GM- and G-CSF in some patients, which was positively correlated with neutrophils in these samples.18,227–230 Although the quality and amount of data varies considerably between diseases, increased in growth factor quantities were generally associated with increased acute damage signals and monocyte and neutrophil chemokines in these patients. Might fibroblasts contribute to the production of these growth factors?
Fibroblasts from various lymphoid and non-lymphoid tissues produce growth factors for myeloid cells and granulocytes, although other cell types including endothelial cells can also play significant roles.66,231 Specialized fibroblasts in the murine lymph node expressed both Csf1/M-CSF and IL-34, the two Csf1r ligands, in vivo and induced expansion of classical monocytes and macrophages in vitro.70,232,233 Fibroblasts in the murine splenic red pulp appeared to serve a similar function as they also expressed both Csf1 and Il34.234,235 Targeted deletion of Csf1 in splenic red pulp, but not white pulp, fibroblasts dramatically reduced red pulp macrophages (Figure 2).235 These data support the importance of fibroblast-derived growth factors for maintenance of myeloid cells during homeostasis. What about after injury?
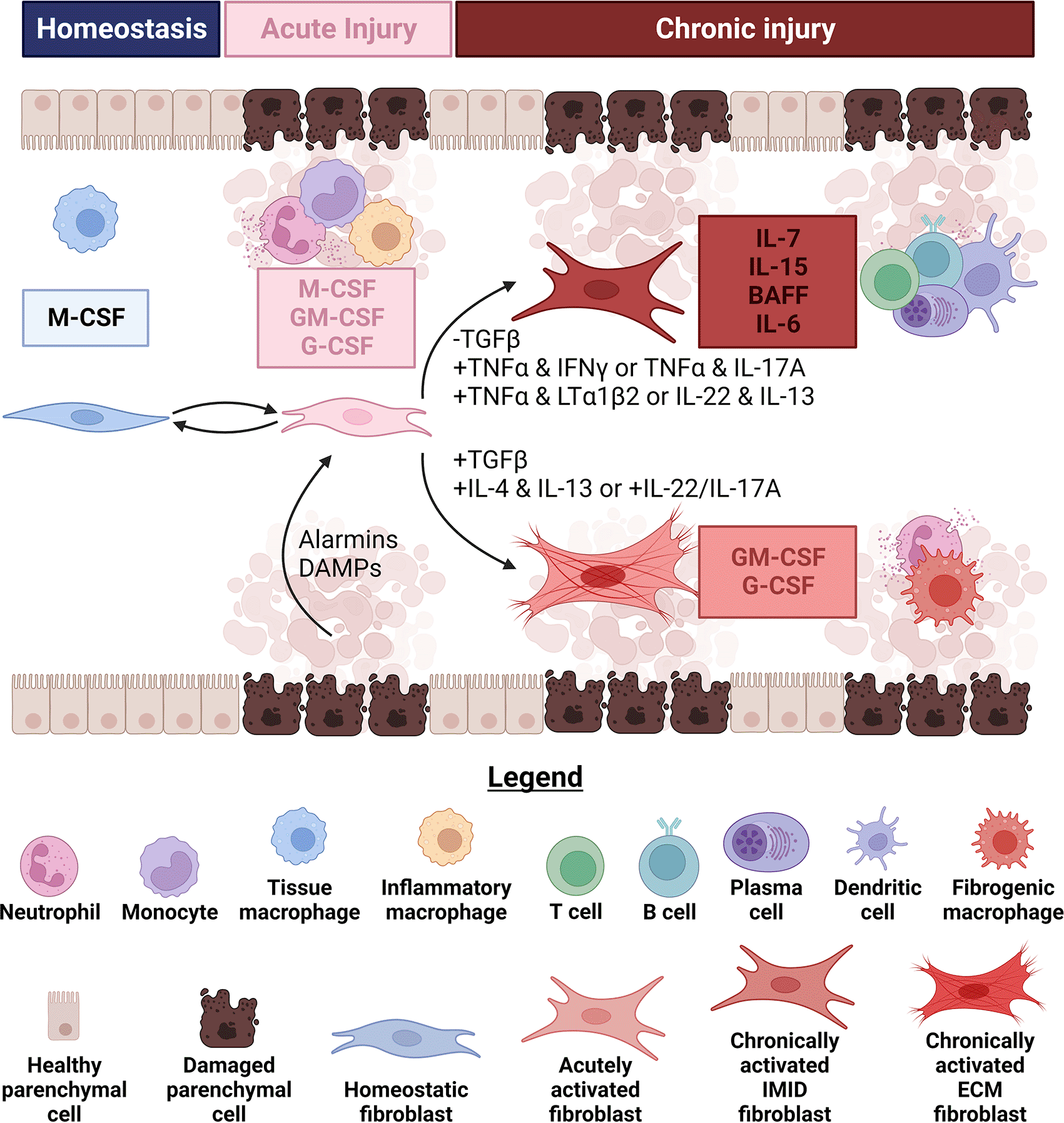
Figure 2. Fibroblasts influence changes in leukocyte infiltrate size and composition by altering the types and amounts of growth factors.
During homeostasis, fibroblasts in several lymphoid and non-lymphoid tissues produce growth factors (generally Csf1/macrophage colony stimulating factor (M-CSF)) to sustain tissue-resident macrophage populations. Immediately upon injury, fibroblasts increase the amount of growth factors they secrete and begin producing Csf2/granulocyte-macrophage colony stimulating factor (GM-CSF) and Csf3/granulocyte colony stimulating factor (G-CSF). Increases in the amounts and breadth of growth factors produced by fibroblasts coincide with their elevated secretion of chemokines that attract granulocytes, monocytes and macrophages (described in subheading 1). Alarmins, damage-associated molecular patterns (DAMPs) and serum from leaking blood vessels can each stimulate these functional changes in fibroblasts. These acute damage signals can also synergize to further enhance the quantities of growth factors produced by fibroblasts. If the injury is successfully contained, fibroblasts can help replenish the depleted tissue-resident macrophage population by attracting monocytes and secreting M-CSF to induce their differentiation. Conversely, if damage signals persist chronic fibrotic (FD) or immune-mediated inflammatory diseases (IMID) can result partly because of divergent fibroblast functional states. Retention of dendritic cells (DCs) and infiltration of type 1 or type 3 polarized αβ memory T cells lead to progressive differentiation of fibroblasts in response to combinations of cytokines like tumor necrosis factor α (TNFα) and interferon γ (IFNγ) or TNFα and IL-17A. DCs can also relax fibroblasts, which decreases contractile activation of latent transforming growth factor β (TGFβ). Mature DCs or activated lymphocytes can produce TNFα and lymphotoxin α1β2 to initiate a second round of fibroblast specialization, triggering secretion of growth factors like IL-7 and IL-15 for T cells and BAFF and IL-6 for B and plasma cells. Fibroblasts appear to reach similar functional states in response to IL-22 and IL-13 in at least one murine model of IMID, although how these cytokines can induce such distinct fibroblast states in FD and IMID is currently unclear. Specialized fibroblasts producing IL-7, IL-15, BAFF and IL-6 likely underpin the formation of large lymphocyte aggregates or tertiary lymphoid structures in IMID. On the other hand, activation of latent TGFβ through several mechanisms in FD, including by contractile fibroblasts, synergizes with IL-4 and IL-13 or IL-22 and IL-17A to maintain production of GM-CSF and G-CSF. Both support monocyte differentiation and granulocyte survival within the fibrotic niche. Figure created with BioRender.
One-time depletion of homeostatic murine macrophages by diphtheria toxin induced a controlled, acute inflammatory response. Csf1 expression was induced in hepatic stellate cells (fibroblastic cells of the liver) upon acute depletion of liver resident Kupffer cells. Repopulation of the Kupffer cell niche by monocytes required M-CSF-Csf1r signaling. In this model, however, Csf1 was expressed by both stellate cells and liver sinusoidal endothelial cells in response to IL-1α, IL-1β and TNFα. The relative importance of Csf1 expression by each cell type remains an open question (Figure 2).66 A similar observation was made after acute depletion of GATA6+ cavity macrophages. Here, both mesothelial cells and capsule fibroblasts provided M-CSF.236 Again, the relative contributions of these two cell types to M-CSF production awaits experimental validation in this model. Regardless of their relative importance, these data indicate that fibroblasts play significant roles in the homeostatic maintenance of tissue macrophage and the restoration of tissue macrophage populations after acute injury. As we saw in the chemokine section above, fibroblasts responded to the alarmins produced in acute injury by attracting both monocytes and granulocytes. Like M-CSF, do fibroblasts supply Csf2/GM-CSF or Csf3/G-CSF to support these infiltrating innate immune cells?
In vivo many different cell types produce GM-CSF. Unlike the M-CSF-dependent tissue macrophages described in previous paragraphs, alveolar macrophages require GM-CSF in both humans and mice.237–244 Multiple cell types expressed GM-CSF in the homeostatic murine lung including type 2 innate lymphoid cells (ILC2), γδ T cells and alveolar type 2 epithelial cells (AT2). However, only GM-CSF from AT2 was required for alveolar macrophage differentiation and maintenance.245 The critical source of CM-CSF may differ between sites, and likely changes in disease. For example, in patients with various forms of inflammatory arthritis fibroblasts, endothelial cells and macrophages all appear to produce GM-CSF.246 To some extent this is recapitulated in the murine SKG arthritis model where GM-CSF production by fibroblasts and ILC2 is necessary for disease, but not in the serum-transfer induced arthritis model where GM-CSF from NK cells is required (Figure 2).247,248 The critical sources of G-CSF in vivo during homeostasis and insult or disease are less clear, although production by endothelial cells is necessary for TLR4-driven emergency granulopoiesis.249 These data indicate that fibroblasts may be important sources of GM- and G-CSF in IMID and FD, but that further work is necessary to determine this in vivo.
In vitro quiescent, growth inhibited human dermal fibroblasts produce almost no leukocyte growth factors. They respond to acute serum stimulation alone (in the absence of alarmins or other exogenous cytokines) by producing mostly bioactive M-CSF (little to no GM- and G-CSF). Stimulation with IL-1α induced secretion of M-, GM- and G-CSF with amounts of M->GM->G-CSF. Acute serum and IL-1α stimulations demonstrated additive synergy in the production of all three growth factors (Figure 2).250 Human lung fibroblasts also secreted M-CSF in response to acute serum stimulation. TNFα stimulation demonstrated additive synergy with serum in the induction of M-CSF, and triggered GM-CSF production. Co-treatment with the corticosteroid dexamethasone inhibited the induction of GM-CSF but not M-CSF by TNFα suggesting a potential mechanism of action for this steroid.251 This growth factor program appears to be conserved as TNFα, IL-1β, IL-17A or IFNγ each increased the ability of synovial, bone marrow and skin fibroblasts to support neutrophil survival in vitro, which largely depended on GM-CSF.90,222,252 Thus, fibroblasts activated by alarmins or serum in FD and IMID may contribute to the local survival of monocytes, macrophages and granulocytes although this may differ by tissue or disease (Figure 2). Further work is necessary to determine how important the production of these growth factors by fibroblasts is and will benefit greatly from the use of genetically engineered mice allowing inducible, fibroblast-specific deletion of these growth factors.
Acute injury signals appear to be shared between FD and IMID. What leads to the expression of differentiated growth factor programs by fibroblasts translate into these divergent clinical manifestations? As acute injuries become chronic diseases, new signals arise with the changing cellular composition and continuing damage. Layering these new signals on top of chronic alarmin, serum and PRR signaling, and the cross-repression between these signals, is what seems to modify the growth factor programs of fibroblasts. This kind of cross-repression is common during organ development and immune cell differentiation.1,67,72,253 Studies of how sequential or simultaneous signals regulate the provision of growth factors by fibroblasts in adulthood are a newer undertaking with many open questions. Investigating these more complex interactions is, however, necessary to understand human diseases which are often characterized by mixed inflammatory and morphogenic signals.115,254
TGFβ is one of the dominant cytokines mediating fibroblast activation and fibrosis in multiple tissues (reviewed in Refs. 255, 256). It is also a potent suppressor of some types of immune responses (reviewed in Refs. 257, 258). Given the relatively small size of leukocyte infiltrates in most FD (compared to IMID), and the predominance of myeloid cells and granulocytes, an obvious question is: does combining TGFβ with pre-existing alarmin, PRR or serum signaling suppresses the production of M-CSF, GM-CSF or G-CSF by fibroblasts? To our knowledge no publications have directly explored this question, but two pieces of evidence suggest that TGFβ may not suppress production of these survival factors. First, TLR4 knockout mice are protected from hepatic fibrosis, which correlates with lower levels of TGFβ signaling in hepatic stellate cells, reduced production of macrophage and neutrophil chemoattractants by hepatic stellate cells and reduced macrophage infiltration into injured livers.118 The reduction in both hepatic stellate cell TGFβ signaling and hepatic macrophage infiltration in TLR4 knockouts argues against TGFβ suppressing the production of myeloid survival factors by HSC. Second, serum or IL-1 alone stimulates the secretion of M-CSF, GM-CSF and G-CSF from human fibroblasts, and the combination of serum plus IL-1 demonstrates additive synergy.250 The 10% FBS-supplemented media that induced secretion of these three CSFs by fibroblasts likely contained ≥1000 pg/mL of bovine TGFβ, which can be activated by freezing and thawing and has bioactivity on human cells.250,259,260 Thus, it seems likely that fibroblasts continue to produce myeloid and granulocyte growth factors to support key constituents of the fibrotic niche when exposed to TGFβ, although this needs to be tested in vitro and in vivo (Figure 2).
What about IMID where many patients have large increases of T and B cells that are frequently found in aggregates? IL-7 protein levels were higher in the synovial fluid of some RA patients compared to OA patients and it was locally transcribed in the synovium.261,262 Levels of IL-15 protein in synovial fluid of RA patients were also higher than OA patients in some cohorts, and this was also transcribed in the synovium.262–265 Fibroblasts isolated from the synovial tissue of RA patients spontaneously secreted more IL-7 and IL-15 than OA synovial fibroblasts. Production of both IL-7 and IL-15 from RA synovial fibroblasts increased in response to IL-1β or TNFα suggesting a role for NF-κB.262,266 IL-7 was detectable in tears from both IgG4-related disease and SS patients, however levels in SS patients did not appear to differ from healthy controls.224–226 IL-7 and IL-15 protein levels were also higher in urine from patients with active lupus nephritis compared to inactive systemic lupus erythematosus, other chronic kidney diseases and healthy controls.223 Do fibroblasts provide these growth factors in IMID to support the observed increases in lymphocytes?
Small aggregates predominantly comprised of T cells are likely sustained by IL-15 presented by myeloid and endothelial cells, IL-2 from locally proliferating T cells and IL-7 produced by lymphatic endothelial cells.91,267–272 Medium and large aggregates, though, appear to require the expansion and differentiation of specialized fibroblasts.14,137,273 Although we currently lack a definitive mechanistic explanation for this observation, specialized fibroblasts likely control the size of T cell aggregates in several ways. Fibroblastic reticular cells (FRC), which are found in the T cell zones of secondary lymphoid organs (SLO), directly control T cell numbers by limiting or increasing survival signals such as IL-7 and IL-15.184,185,269,274–276 The mechanisms by which fibroblasts are activated to produce IL-7 and IL-15 are still unclear. Genetic ablation of LTβR in Ccl19+ lymph node fibroblasts reduced Il7 expression, and transgenic overexpression of LTβR ligands in the pancreas induced Il7 suggesting that LTβR signaling may be sufficient.184,192 However, pharmacologic or genetic LTβR antagonism increased Il7 transcripts in bone marrow fibroblasts.277
Fibroblast regulation of T cell numbers in primary (PLO) or SLO through production or restriction of growth and survival factors like IL-7 seems likely.184,185,274,278–280 Expansion of specialized fibroblasts that produce similar T cell survival factors is likely required to increase the carrying capacity of inflamed tissues and support medium and large aggregates (Figure 2).14,105,107,185,191,252,268,281 However, despite our presentation of circumstantial evidence for this hypothesis it has yet to be directly tested. Rigorously testing this hypothesis is challenging as fibroblasts express similar transcriptional programs across tissues, and tissue-specific fibroblast modules are often shared with non-fibroblast cells in those same tissues.70,174,190,282 Conditional deletion is clearly necessary to avoid confounding developmental defects, as seen in the constitutive, fully-body IL-7 knockout.283 To untangle effects specific to the inflamed non-lymphoid tissue from potential effects of growth factor deletion in PLO or SLO fibroblasts, we propose orthotopic adoptive transfer of purified fibroblasts from mice genetically modified for conditional growth factor deletion or parabiosis between wild type and mice with fibroblast-specific conditional growth factor deletions.105,284 These experiments could be complemented by spatial transcriptomics or combined in situ hybridization and protein staining in biopsies from human inflammatory diseases to show neighborhood-based enrichment of IL-7 expression specifically by fibroblasts scaffolding large T cell aggregates. Testing for cross-species conservation is critical given the differences in production and presentation of growth and survival factors (e.g. IL-15) and fibroblast behavior between species.14,270–272,285,286
Specialized fibroblasts in primary and secondary lymphoid organs are major producers of the B cell growth factors IL-6, BAFF and APRIL, as well as the marginal zone B cell survival factor Delta-like 1.70,233,287–291 In fact, BAFF is expressed by multiple fibroblast subsets in lymph nodes: one within B cell follicles, one in the interfollicular T cell zones and one in the medullary cords.233,287,292,293 These microanatomically distinct fibroblasts are also functionally different with some supporting B cells and others supporting plasma cells.287,293 Increased BAFF expression has been detected in the affected joints of RA patients and in the affected glands of SS patients.107,294 For both RA and SS patients with lung complications, increased BAFF expression is also detected in patients with pulmonary B cell-containing lymphoid aggregates.21 Local production of survival factors appears especially important to support autoreactive B cells in IMID. Autoantigen-specific B cells are particularly sensitive to activation-induced death due to chronic B cell receptor (BCR) stimulation, and BAFF can rescue these autoreactive B cells through non-canonical NF-κB-mediated Pim2.295 Simply observing increased production of B cell growth factors in IMID does not necessarily mean these are being produced by fibroblasts, though. Myeloid cells and granulocytes are also capable of producing many of these proteins.296–300 What evidence supports these factors coming from activated fibroblasts?
There are two lines of evidence. First, specialized fibroblasts producing BAFF transcripts have been identified by scRNAseq in dissociated joints from RA patients and salivary glands from SS patients (Figure 2).107,189 Second, synovial fibroblasts isolated from RA patients secrete BAFF in response to TLR3 or TNFα stimulation and constitutively secrete APRIL.143,301 Matched dermal fibroblasts from the same RA patients also produced BAFF transcripts in response to TLR3 and TLR4 stimulation, but secreted ~10-fold less BAFF and APRIL than synovial fibroblasts even after stimulation.301 Production of these growth factors by fibroblasts is effective as synovial fibroblasts support B cell survival in vitro in co-cultures.302 Whether BAFF expression increases prior to B cell infiltration as a prerequisite for B cell infiltration, or pioneer B cells induce BAFF to create a feed-forward loop is an open question. Based on kinetic studies in a murine model of SS where elevation of BAFF transcripts appears slightly before B cell infiltration we propose that early alarmin signaling by tissue resident lymphocytes is necessary to induce TLR3/4 and/or TNFR2 expression by fibroblasts. In turn, activation of tissue resident lymphocytes, either by fibroblasts or other cells in close proximity, induces surface expression of TNFα or LTα homotrimers to trigger TNFR2-driven non-canonical NF-κB activation and BAFF transcription in fibroblasts (Figure 2).107,153,154,196,301,303 These data suggest a tissue- or inflammation-driven epigenetic and post-transcriptional regulation program that differentiates RA synovial and dermal fibroblasts, and may partly account for the accumulation of B cells in affected joints but not unaffected skin of these patients.
Together, these data suggest that fibroblasts may provide critical growth factor support for leukocytes infiltrating tissues affected by FD and IMID. In these tissues, fibroblasts appear to adopt distinct activation or differentiation states that evolve as acute insults become chronic diseases with distinct clinical manifestations. This increasing fibroblast specialization seems to result from progressive signaling dialogues that differ between fibrotic diseases and IMID. Provision of specific collections of growth factors by IMID or FD fibroblasts may be a result of these different combinations of signals and a mechanism to control the size and composition of leukocyte infiltrates. We propose that this is necessary for the different clinical manifestations and organs affected by various FD and IMID. Our theory is that cross-repression and synergistic induction of receptors and genetic modules in fibroblasts drive these distinct fibroblast functions, pathology and tissue tropism in IMID and FD.
Masters of the extracellular matrix (ECM)
When thinking about the pathogenic functions of fibroblasts, the most obvious starting point is fibrosis. Fibrosis, or scarring, is the result of abnormal repair in response to chronic injury. The molecular and cellular mechanisms that cause fibrosis are complex, but removal of healthy, homeostatic ECM and its replacement by pathologic fibrillar collagen is a major part of these diseases. Fibroblasts are at the center of this process, and what usually comes first to mind is the myofibroblast. Conventionally, the definition of a myofibroblast includes two components: production of fibrillar collagens and other ECM proteins, and contractility which is usually shorthanded to α-SMA+ fibroblasts (reviewed in Ref. 304).134 We agree with the long-form definition of “myofibroblast”. However, activated fibroblasts that lack detectable α-SMA protein express fibrillar pro-collagen proteins and are associated with areas of fibrosis in some diseases.305 Thus, we prefer the more general term “ECM fibroblast” when referring to activated fibroblasts in FD with myofibroblasts being a subset of ECM fibroblasts.
Fibrosis can occur in chronic diseases with either type 2 or type 3 immune polarization (or in the absence of obvious inflammation or leukocyte infiltration), but this clear-cut skewing is more common in animal models than patient cohorts. More frequently, human fibrotic diseases demonstrate varying degrees of mixed inflammation. Due to cross-repression and rebound inflammation, stopping progressive scarring in patients with obvious inflammatory skewing will likely require simultaneous antagonism of both type 2 and type 3 signaling.115,116,254,306 Even more complicated, the predominant inflammatory skewing of FD patient cohorts likely varies depending on the organ and stage of disease in the biopsy. For example, the lungs of active IPF patients generally have three distinct zones: relatively healthy, inflamed with little fibrosis and overtly acellular due to end-stage fibrosis (reviewed in Ref. 307). Depending on the zone sampled researchers will get wildly divergent results. Although this example is true for IPF, similar caution is warranted when interpreting findings from other fibrotic diseases. The extent and predominance of type 2, type 3 or a lack of overt inflammation among patients with different fibrotic diseases is an open area of research requiring larger patient cohorts of diverse genetic backgrounds.
IL-4, IL-5 and IL-13 are the canonical cytokines associated with type 2 inflammation.308 Increased levels of these cytokines have been detected in patients with a variety of sterile fibrotic diseases including NASH and IPF. Depending on the cohort and the way in which the measurements were taken, higher levels of these cytokines are associated with more severe and/or longer disease duration.115,254,309–311 Fibroblasts isolated from the lungs of IPF patients express more of the IL-4 receptor α (IL-4Rα) and IL-13 receptor α1 (IL-13Rα1) than fibroblasts from healthy lungs. These two proteins form the heterodimeric type II receptor for IL-4 and IL-13, and this elevated expression was associated with increased COL1 transcription in response to these cytokines.312,313 IL-4 and IL-13 can also stimulate COL1 secretion by some human hepatic stellate cell lines.314 However, the mechanisms by which these cytokines, especially IL-13, induce fibrosis may differ between organs or diseases.
Liver fibrosis induced by local transgenic overexpression of IL-13 and signaling through the type II receptor on fibroblasts (including hepatic stellate cells) may be independent of TGFβ.116,315,316 Pulmonary fibrosis induced by similar transgenic overexpression of IL-13 in murine lungs, although induced using a different genetic technique, required TGFβ for fibrosis (Figure 3A).317,318 Why this distinction was observed is currently unclear. Different results were obtained from the strongly type 2 polarized Schistosoma mansoni models of lung and liver fibrosis, though, with both appearing to be independent of TGFβ.116,316 Perhaps ECM fibroblast activation and fibrosis in these models depends on the magnitude of the IL-13 response or the balance between mixed inflammatory polarization?
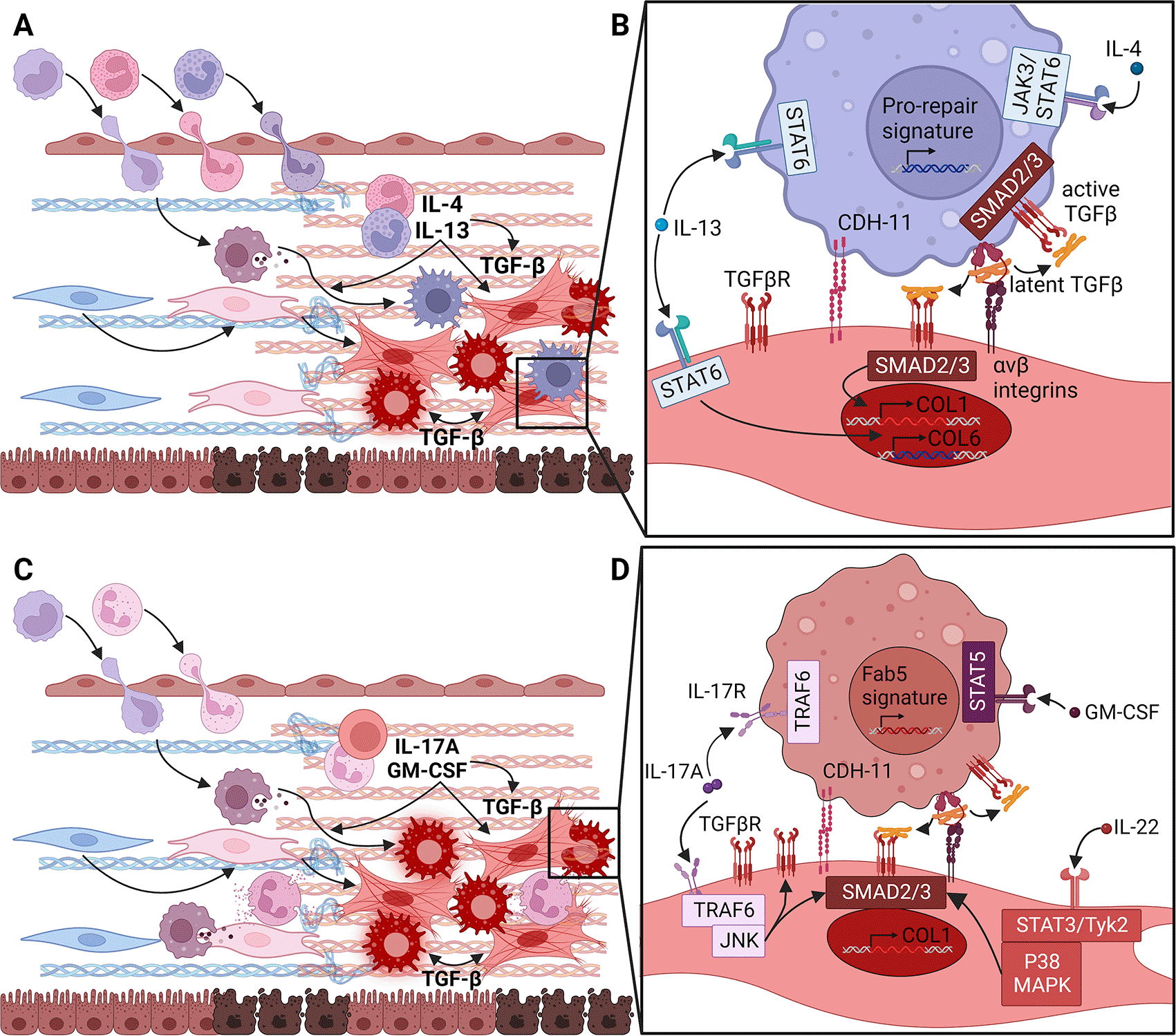
Figure 3. Type 2 or type 3 cytokines can differentiate extracellular matrix (ECM) fibroblasts, which form distinct fibrotic niches.
(A) In predominantly type 2 skewed fibrotic diseases (FD), acutely activated fibroblasts and other cells attract monocytes, eosinophils and basophils. Degranulation of eosinophils and basophils can release IL-4 and IL-13 to activate the transcription factor STAT6. Eosinophils granules can also release TGFβ to promote differentiation of ECM fibroblasts into myofibroblasts through the transcription factors SMAD2 and SMAD3. IL-4 and IL-13 also influence monocyte differentiation into macrophages, which can then add to local TGFβ production and myofibroblast differentiation. (B) By signaling through the type II receptor, IL-4 and IL-13 can directly activate production of Collagen type VI (COL6) by fibroblasts. Thus, they may directly drive fibrosis without relying on TGFβ although in this situation it is unclear how production of fibrillar collagens is initiated. IL-4 and IL-13 can also stimulate TGFβ production by macrophages. Thanks to homotypic Cadherin-11 (CDH-11) synapses formed with myofibroblasts, the TGFβ produced by these macrophages can be converted from its latent to active form through fibroblast integrin αV heterodimers and contractile force. Thus, IL-4 and IL-13 may trigger Collagen type I (COL1) production by ECM fibroblasts through this indirect TGFβ pathway. (C) In FD dominated by type 3 inflammatory skewing, acutely activated fibroblasts and other cells primarily induce monocyte and neutrophil extravasation. Under these circumstances, γδ T cells, neutrophils and mast cells produce IL-17A and, with fibroblasts, GM-CSF. These induce the differentiation of infiltrating monocytes into Fab5 macrophages. IL-17A can also potentiate low levels of TGFβ signaling generated through contractile activation by ECM fibroblasts expressing unbound podoplanin (PDPN). (D) GM-CSF produced by fibroblasts and other cells initiates monocyte differentiation towards the Fab5 macrophage state through the transcription factor STAT5. As described in (B), Fab5 macrophages can also form CDH-11 synapses with ECM fibroblasts and provide latent TGFβ for activation by fibroblast integrin αV and contraction. Active TGFβ can augment or stabilize the Fab5 program in macrophages and promote ECM fibroblasts to become myofibroblasts. IL-17A potentially produced by several cell types in the fibrotic niche can increase expression of TGFβRII and enhance SMAD2 and SMAD3 activation by signaling through TRAF6 and JNK in fibroblasts. In differentiating monocytes, IL-17A can also promote differentiation into Fab5 macrophages. IL-22, another type 3 cytokine, can also enhance SMAD2 and SMAD3 activation in concert with TGFβ. Figure created with BioRender.
In a murine model of severe asthma characterized by strong, mixed type 2 and type 3 inflammation neither reducing TGFβ activation nor IL-13 blockade alone or in combination decreased fibrosis.116 This last observation suggests two possibilities. First, that in the presence of multiple strong, non-redundant ECM fibroblast activating signals the degree of combined inhibition achieved insufficient target coverage. Second, that this model drives ECM fibroblast activation by an additional, orthogonal mechanism. Regardless of which is true, these data clearly demonstrate that type 2 inflammation, and specifically IL-13, can induce ECM fibroblast activation and fibrosis both directly and indirectly through TGFβ. One other part of this story is curious: IL-13 appears to directly stimulate Col6a1 transcription via STAT6 activation in fibroblasts, but the mechanism by which IL-13 induces COL1 production is unclear (Figure 3B).315 Collagen type VI is not a traditional fibrillar collagen like types I, II and III but instead forms beaded microfilaments that are assembled into reticular structures (reviewed in Ref. 319). Perhaps dramatic increases in local Collagen type VI stiffen the local matrix and cascade through outside-in integrin signaling and activation of latent growth factors like TGFβ?
As noted above, type 3 inflammation is also associated with fibrosis. Type 3 inflammation is characterized by the recruitment and activation of T-helper (Th) 17 cells, γδ T cells, type 3 innate lymphoid cells (ILC3) and neutrophils (Figure 3C). Infiltrates where these cells predominate plus local activation of mast cells leads to the secretion of IL-17A, IL-22 and GM-CSF (Figure 3C).254,320–322 Type 3 immune responses have been associated with fibrosis and cancer progression in many organs. IL-17A cytokine or receptor knockout animals are protected from fibrosis in variety of pre-clinical models.72,309,323 Interestingly, opposing pro- vs anti-fibrotic functions have been reported for IL-22 depending on timing, type of injuries and opposite effects reported on epithelium vs mesenchymal cells.323–326 Increased expression of IL-17A is associated with IPF, NASH and other fibrotic diseases while, to our knowledge, increased IL-22 has only been observed in NASH patients.254,309 Elevated GM-CSF expression is also associated with NASH, IPF and mustard gas pulmonary fibrosis.229,230 As we have described in previous sections, type 3 cytokines are associated with recruitment and survival of macrophages and granulocytes in FD, but also with the recruitment of DCs and lymphocytes in IMID. Do type 3 cytokines drive similarly opposing ECM depositing and eroding functions in fibroblasts?
IL-17A is the prototypical type 3 cytokine, and its receptor is expressed on a variety of cell types including epithelium, endothelium, mesothelium, and myeloid cells.327 It signals through a heterodimeric receptor composed of the IL-17RA and IL-17RC subunits triggering a cascade through the Act1/TRAF6 to activate NF-KB, ERK1/2, C/EBPB, JNK and MAPK pathways (reviewed in Ref. 328). In the absence of serum, IL-17A only triggers secretion of pro-inflammatory chemokines in primary human hepatic stellate cells. In contrast, IL-17A sensitizes myofibroblasts to the action of TGFβ by stabilizing its receptor on the surface of cells in a JNK-dependent manner (Figure 3C).329 In conclusion, IL-17A is pleiotropic cytokine which impacts myofibroblast directly and/or indirectly within the fibrotic niche.
IL-22 is a complex pleiotropic cytokine from the IL-10 family, with opposite roles in health and disease. IL-22 signals through a heterodimeric receptor composed of the common IL-10 family chain IL-10RB and IL-22RA1 triggering activation of STAT3 and MAPK.330,331 Expression of the IL-22 receptor complex is restricted to epithelial cells and fibroblasts. IL-22 promotes the survival and proliferation of epithelial cells via STAT3 promoting tissue repair and therefore limiting fibrosis.330 On the other hand, IL-22 signaling also induces the secretion of chemokines involved in the recruitment of neutrophils, monocytes, and macrophages.332,333 Studies in the liver also demonstrated that IL-22 contributes to fibrosis progression by enhancing TGFβ signaling via STAT3/MAPK activation (Figure 3D).254 While IL-22 deficient mice have impaired tissue repair mice lacking the decoy receptor IL-22BP also have impaired tissue repair due to aberrant inflammatory responses. These discrepancies require further investigation but likely depend on the levels of IL-22 and IL-22BP, the timing of IL-22 production, the cells receiving IL-22 signals and the other signals being simultaneously received and integrated.
Pathogenic production and organization of the ECM in fibrotic diseases is clearly one function of fibroblasts. Do they serve similar, but opposite, roles in inflammatory diseases characterized by excessive destruction of the ECM like RA? The answer appears to be yes, which is especially important when considering treatment for rheumatoid arthritis patients with the fibroid pathotype and osteoarthritis (OA) patients. Due to the low frequency of lymphocytes, myeloid cells and granulocytes in the joints of these patients next generation RA drugs should simultaneously block multiple pathways to cover patients with all three pathotypes.161 Given the similarities between the fibroid RA pathotype and OA, these drugs may work for both groups of patients.
In rheumatoid arthritis mouse models, PDPN+FAPα+THY1- synovial fibroblasts specialize in joint destruction and extracellular matrix remodeling as demonstrated through engraftment. They secrete high levels of matrix metalloproteinases (MMPs) 3, 9 and 13, which are especially effective at degrading the most abundant fibrillar collagen in cartilage, Collagen type II, and the major constituent of basement membranes, Collagen type IV.171,284,334 These PDPN+FAPα+THY1- synovial fibroblasts described by Croft et al. are likely the same as the PRG4+ synovial fibroblasts described by Micheroli et al. Both lack or have undetectably low expression of THY1, express MMP3 and appear to be associated with the destruction of synovial cartilage independent of leukocyte infiltrates.284,335
What activates these destructive functions in fibroblasts? IL-1, IL-17A, TNFα and Oncostatin M all induce combinations of MMPs in synovial fibroblasts, which result in degradation of glycosaminoglycans or cartilage in vitro.144,336,337 Connecting MMP gene or protein expression with functional consequences is particularly important given the tight regulation of their activity. Pro-MMPs undergo proteolysis before becoming catalytically active, and after activation MMPs can be inhibited by the tissue inhibitor of metalloproteinases (TIMP) family of proteins (reviewed in Ref. 338). The balance between protease production, activation and suppression is also relevant for ECM fibroblasts. Clearance or modification of homeostatic ECM often accompanies the deposition of fibrotic ECM, and this requires the activation of specific classes of proteases and the inhibition of others. Although it is beyond the scope of this review, the generation of matricryptins or matrikines (proteolytic fragments of ECM molecules that induce signaling distinct from their larger, intact parent molecules) likely play roles in both fibrotic diseases and IMID (reviewed in Refs. 319, 339, 340). Despite the complexities of studying the pathogenic destruction of ECM by fibroblasts, these data indicate that fibroblasts are sufficient for clinically relevant joint destruction. Further research is necessary to determine whether fibroblasts make significant contributions to ECM destruction in other diseases.
It may be surprising that some of the type 3 cytokines associated with pathologic deposition and organization of ECM by fibroblasts can also induce the opposite outcome. For example, why is IL-17A associated with fibrosis in murine models and human NASH and IPF patients and with joint destruction in murine models and human RA patients? This is still an open question. However, these distinct pathologies and disease outcomes may be due to different signals combining with IL-17A in fibrotic versus eroding diseases. In FD, the combination of IL-17A and TGFβ signaling in fibroblasts can potentiate more robust ECM gene expression than either cytokine alone.329 Add outside-in sensing of the stiff fibrotic matrix and hypoxia on top of the synergy between IL-17A and TGFβ, and this combination is likely part of what drives fibroblast to deposit pathogenic ECM in FD. There is less TGFβ and more TNFα signaling in RA to combine with IL-17A, which results in higher MMP13-driven proteoglycan destruction.179,336 Similarly, how does IL-13 promote ECM fibroblast differentiation and fibrosis either alone or in combination with TGFβ in some models, while also driving fibroblast activation in TLS formation in a model of SS?
These data clearly demonstrate that distinct fibroblast activation or differentiation states and functions occur in FD and IMID. ECM fibroblasts are at the heart of FD. They are the major producers and organizers of collagens and many other ECM molecules, and tune the stiffness of tissues. By modifying the ECM in this way, fibroblasts also activate latent growth factors and convert haptotactic to chemotactic gradients. Are all ECM fibroblasts the same, or are there different functional states that arise from predominant type 2 versus type 3 activation (or in the absence of detectable inflammation)? And do all fibrotic fibroblasts deposit the same ECM constituents or exert the same contractile forces, or is collaboration between multiple ECM fibroblast states necessary for fibrosis? Answering these questions will be necessary for the rational development of therapeutics that stop or even reverse FD. For example, if the fibroblasts activated by type 3 cytokines build fibrotic ECM with a different composition from those activated by type 2 cytokines the molecular pathways requiring therapeutic manipulation will differ. This would also necessitate the development of companion diagnostics to effectively select patient populations for clinical trials and deliver the most effective personalized medicine to FD patients.
Conversely, different fibroblast subsets are responsible for ECM destruction or lymphocyte recruitment and support in RA. Thus, multiple fibroblast functional states can coexist and collaborate to drive complex disease pathology. Is ECM degradation by erosive fibroblasts necessary for the expansion of fibroblasts that scaffold lymphocyte aggregates and TLS? As thickening and stiffening of the lymph node capsule appears to be a compensatory reaction that constrains swelling and limits lymphocyte proliferation, some degradation of the local ECM is likely necessary for the growth of lymphocyte aggregates.108,109,139 That erosive fibroblasts are sufficient for destruction of the collagen type II-rich cartilage in synovial tissues also begs the question of how important different cell types are to to ECM destruction. Similarly, if erosive fibroblasts in RA can significantly destroy the synovial matrix do they also make major contributions to removal of the healthy, homeostatic matrix in fibrotic diseases? Addressing these questions is critical to developing better therapies for patients with both erosive IMID and FD. Understanding the signals that create erosive versus ECM fibroblasts and whether these are stable differentiation states or reprogrammable activation states will help us develop the appropriate therapeutic regimens to treat these opposing disease pathologies.
Subheading 4: Controlling leukocyte differentiation and activation in non-lymphoid tissues
Up to this point we have demonstrated that fibroblasts appear to adopt progressive, functionally distinct activation states in IMID versus FD. In IMID these various activated fibroblasts produce chemo-/haptotactic and growth factors specialized to support local adaptive immune responses. Conversely, in FD ECM and myofibroblasts generally attract smaller infiltrates of predominantly macrophages and granulocytes through specialized chemo-, hapto- and durotactic gradients and growth factors for these cells. Fibroblasts can also exert significant, opposing effects in these diseases by depositing or, at least in RA, degrading large amounts of ECM. The final question we will tackle is: do specialized fibroblasts contribute to the activation or differentiation of infiltrating leukocytes in these diseases?
Monocytes and macrophages are important parts of both IMID and FD, although they are a much larger proportion of infiltrates in FD than IMID. Recently, several reports identified scar-associated pro-fibrotic macrophages in human and mice in a variety of tissues.17,341–350 A common signature (Fab5) using the combination of CD9, CD63, FAPB5, GPNMB and SPP1 was recently suggested to accurately distinguish these fibrotic macrophages from populations with other functions. Fab5 macrophages originate from monocytes and, in some settings, require IL-17A, GM-CSF (likely from neutrophils, although fibroblasts and other cell types may play a role) and TGFβ for their differentiation in vitro and in vivo (Figure 3D).18,245,247,248 Another route to a Fab5-like macrophage may be through M-CSF and IL-6, both of which can be produced by fibroblasts although the authors of this study identified an autocrine loop of these factors.351 Fab5 macrophages elicit several key pro-fibrotic functions that were elucidated in vitro. Fab5 macrophages contribute to the removal of normal ECM through MMP activity and enhance deposition of fibrillar collagen by ECM fibroblasts (Figure 3D). Although the molecular mechanisms by which Fab5 macrophages activate ECM fibroblasts are actively being studied, they likely include proximal presentation of latent TGFβ for activation by fibroblasts, as well as Amphiregulin and possible TWEAK and PDGFR ligands.17,129,352 The relative importance of ECM fibroblast-produced M-CSF, GM-CSF, IL-6 and activation of latent TGFβ to Fab5 differentiation requires experimental testing, although it seems likely they contribute (Figure 4A).
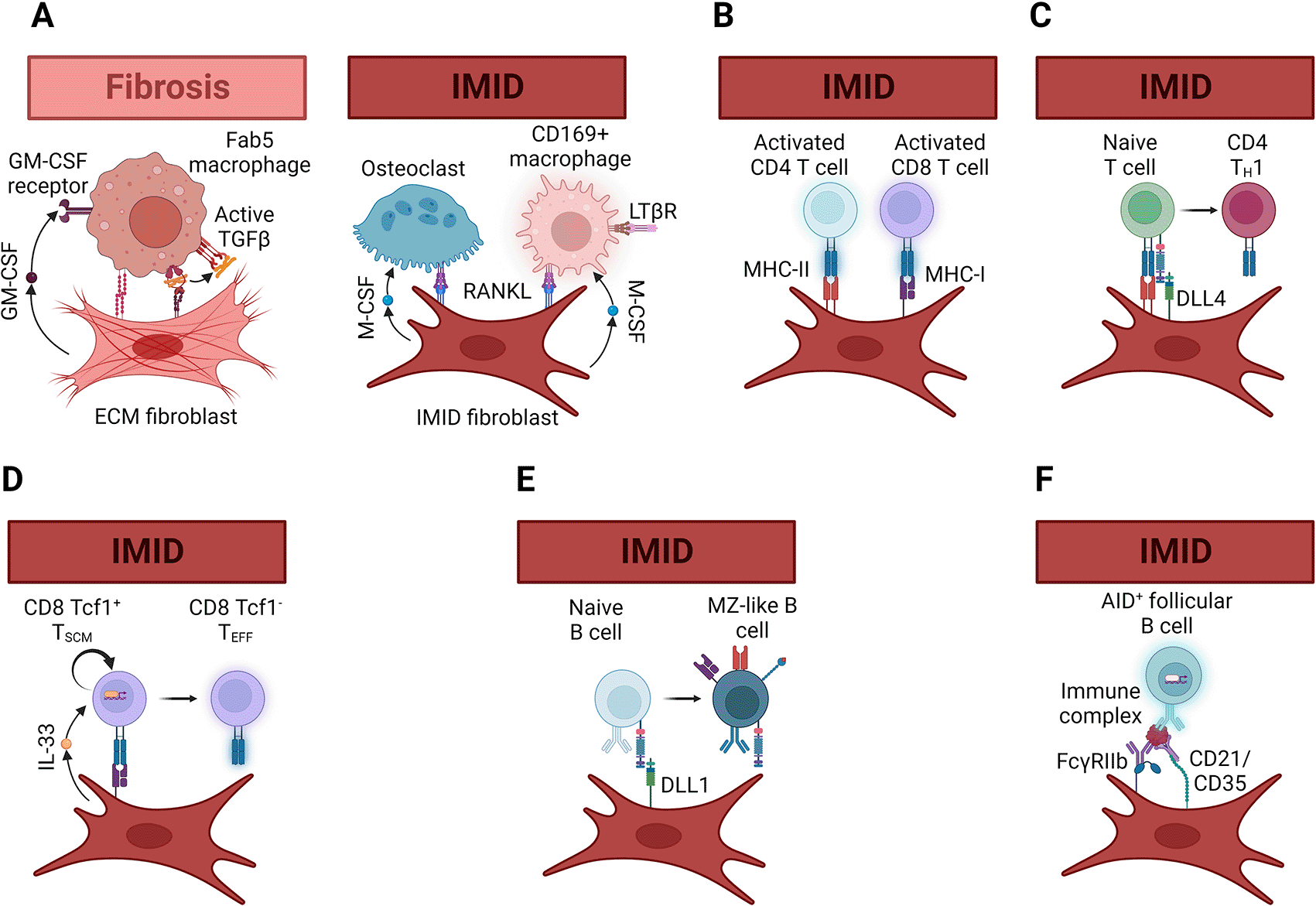
Figure 4. Fibroblast mechanisms to activate leukocytes in fibrotic and immune-mediated inflammatory diseases.
(A) In fibrotic diseases, fibroblasts and macrophages can form Cadherin-11 mediated synapses. By secreting granulocyte-macrophage colony stimulating factor (GM-CSF) and activating latent transforming growth factor β (TGFβ) through integrin αV contractile forces, extracellular matrix (ECM) fibroblasts can promote differentiation of monocytes into fibrotic Fab5 macrophages. On the other hand, in immune-mediated inflammatory diseases (IMID) production of receptor activator of nuclear factor κB ligand (RANKL) and macrophage colony stimulating factor (M-CSF) can lead to osteoclast or CD169+ macrophage differentiation. Although the molecular mechanisms that dictate whether a macrophage becomes and osteoclast or CD169+ macrophage require further investigation, the dependence of CD169+ macrophage differentiation on LTβR signaling may be one differentiating factor. (B) Fibroblasts can express both major histocompatibility class-I (MHC-I) and -II (MHC-II), which can reactivate memory CD8 and CD4 T cells as they infiltrate tissues affected by IMID. Although not discussed in this article, MHC expression by fibroblasts can suppress T cell proliferation or promote tolerance in addition to restimulating T cells429. (C) Fibroblasts in secondary lymphoid organs produce the Notch1/2 ligands Delta-like 1 (DLL1) and 4 (DLL4). In murine models of graft versus host disease, expression of DLL4 and, to a lesser extent, DLL1 by lymphoid organ fibroblasts was necessary for TH1 differentiation of grafted naïve T cells. This DLL1/4-Notch1/2-induced T cell differentiation was required for disease induction although it is unclear whether non-lymphoid organ fibroblasts can perform the same function. (D) IL-33 production by Ccl19+ is necessary for both maintenance of the Tcf1+ CD8 stem memory T cell pool and for effective anti-viral and anti-tumor CD8 T cell differentiation. The mechanism by which IL-33 produced by fibroblasts promotes type 2 immunity in parasite infections and type 1 anti-viral and anti-tumor immunity is unclear, but likely occurs through crosstalk with other signals like type 1 interferons. (E) DLL1-Notch2 signaling induces the differentiation and maintenance of splenic marginal-zone B cells. A similar system may work in tissues affected by IMID, but this awaits experimental testing. (F) Follicular dendritic cells in secondary lymphoid organs trap antibody- and complement-containing immune complexes using the Fcγ receptor IIb (FcγRIIb) and complement receptors 1 (CD35) and 2 (CD21). Presentation of the antigens in these immune complexes to B cells is important for germinal center reactions. Affinity maturation and class-switch recombination mediated by activation-induced cytidine deaminase (AID) in follicles both require this follicular dendritic cell function. Fibroblasts expressing FcγRIIb, CD35 and CD21 appear in the affected tissues of some IMID patients, and stimulation of fibroblasts isolated from these tissues can induce expression of these three receptors. Thus, it is likely that follicular dendritic-like cells can maintain germinal center reactions in IMID tissues. Figure created with BioRender.
Fibroblasts likely induce differentiation of functionally distinct macrophage subsets in IMID. In SLO, differentiation of CD169+ sinusoidal and metallophillic (or marginal zone) macrophages requires Receptor activator of nuclear factor-κB ligand (RANKL/TNFSF11) expression by fibroblasts (Figure 4A). These macrophages capture free virus and initiate anti-viral CD8 T cell responses.353–355 The roles of CD169/SIGLEC1+ macrophages in IMID are unclear. One report on RA patients proposed a protective role for CD169+ macrophages, while others noted a positive correlation of circulating CD169+ myeloid cells with worse and extra-glandular disease in SS and dermatomyositis.356–358 Given the recent reports on potentially pathogenic CD8 T cells, and reductions in synovial CD8 T cells being associated with clinical responses to rituximab in RA patients, we favor the hypothesis that CD169+ macrophages are pathogenic in IMID.161,171,359–363
Several fibroblastic lineage cells in the bone and bone marrow induce osteoclast differentiation, thus indirectly promoting bone and cartilage resorption, by producing RANKL.364,365 Do fibroblasts play similar roles in IMID-associated osteoporosis? PDPN+FAPα+THY1- synovial fibroblasts in RA patients and murine models produce RANKL, which promotes macrophage differentiation into osteoclasts and is associated with bone erosion.171,284,334,366 Although RA appears to be the only IMID directly associated with bone erosion or osteolysis, many IMID are treated with glucocorticoids and long-term use of these steroids is a risk factor for osteoporosis (reviewed in Ref. 367).368 Glucocorticoids can directly induce RANKL expression by fibroblastic cells from several tissues in mice and humans as the glucocorticoid receptor directly binds to the RANKL promoter.369,370 Osteolysis also occurs in adverse responses to joint replacement and is frequently accompanied by histologic macrophage activation.371,372 Both RANKL and its receptor, RANK, are expressed in tissues containing wear particles and osteoclasts spontaneous differentiated in patient samples with high RANKL to Osteoprotegrin (OPG, the soluble decoy receptor for RANKL) transcript ratios.373 Titanium particles were sufficient to indirectly induce RANKL expression by synovial fibroblasts, as was combined stimulation with TNFα and IL-17A which worked through trans IL-6 signaling (Figure 4A).374,375 These data indicate a surprisingly wide range of osteolytic pathologies that appear to be driven by fibroblast-induced osteoclastogenesis.
Do fibroblasts participate in the local re-activation of T cells after they extravasate into inflamed tissues? Fibroblasts (both human and murine) can express Class I (MHC-I) and Class II (MHC-II) Major Histocompatibility Complex molecules.70,171,189,376–384 Murine fibroblasts can activate both CD4 and CD8 T cells in an MHC-dependent fashion to elicit cytokine production and cytotoxic activity (Figure 4B).377,378,380,383–385 Human fibroblasts may be able to do the same, but definitive proof awaits experimental validation.379,382 This could be demonstrated in vitro by obtaining donor-matched fibroblasts from skin biopsies and circulating T cells from the blood, pulsing the fibroblasts with a cocktail of common viral T cell epitope peptides and testing for proliferation and cytokine production. Given the suppression of effector T cell activation by primary human SLO fibroblasts and lymphocytic choriomeningitis virus clone 13 (LCMV-C13) infected murine fibroblasts, blocking co-inhibitory molecules either alone or in combination with CRISPR-mediated knockout of other suppressive factors may be necessary to detect much cytotoxic activity.385–390 The relative importance of MHC expression by fibroblasts, macrophages and DCs in both SLO and IMID also await direct experimental validation. Is antigen presentation the only mechanism by which fibroblasts can restimulate infiltrating T cells?
No, although the relative importance of antigen-specific and -independent reactivation of T cells infiltrating inflamed, non-lymphoid tissues requires further investigation. Fibroblasts may reactivate infiltrating T cells through a variety of antigen-independent mechanisms including cytokine production, expression of canonical or non-canonical co-stimulatory molecules and adhesion proteins. SLO fibroblasts are important sources of the Notch ligands Delta-like 1 (Dll1) and 4 (Dll4) in both homeostasis and graft-versus host disease (GVHD, Figure 4C).281,290,391,392 Homeostatic human dermal fibroblasts lack Notch ligands, but this may change (and not just in the skin) during inflammation as non-lymphoid fibroblasts adopt SLO-fibroblast-like qualities and awaits experimental testing.14,105,393 If true, inflamed fibroblasts expressing Dll1 or Dll4 may reactivate infiltrating T cells to promote inflammatory or autoimmune tissue destruction (Figure 4C).290,391 Expression of Dll1 or Dll4 by inflamed fibroblasts may also promote local tertiary lymphoid tissue formation by driving differentiation of infiltrating naïve T cells into T follicular helpers and naïve B cells into germinal centers.290,394 An alternative explanation is that Dll1 or Dll4 expression is truly restricted to SLO fibroblasts and is only important for imparting the ability to home to inflamed tissues, not the restimulation of tissue destructive effector functions.392 It seems plausible, at least, that activated fibroblasts in IMID can contribute to inflammatory tissue destruction by reactivating infiltrating T cells and driving T cell differentiation via Dll1 and Dll4. This is especially important when considering the translatability of findings from insult-naïve, specific pathogen free mice to adult humans and other animals that have substantial pools of tissue-resident lymphocytes and have epigenetic memories of these past insults.
DLL/Notch signaling is, however, just one of many tools fibroblasts possess to restimulate infiltrating T cells. Fibroblasts in both SLO and non-lymphoid tissues express alarmins like IL-33 and Thymic Stromal Lymphopoietin (TSLP).67,68,71 IL-33 produced by SLO fibroblasts is important for activation and effector differentiation of alloreactive CD4 T cells in murine GVHD models and effector CD8 T cells in acute and chronic viral infection.68,71 Recent findings have pointed to the importance of IL-33 in balancing between CD8 stem memory T cell expansion and effector differentiation in chronic viral infections and cancer (Figure 4D).69,395 In non-lymphoid tissues, IL-33 expressing fibroblasts appear to be enriched at interfaces between microanatomic compartments. These interfaces include airways and vasculature in the lungs, portal triads and vasculature in the liver, vasculature in adipose tissue, hair follicles and subcutaneous fascia in murine skin and the dural sinus in the meninges.67,72 Successful cancer immunotherapies create specialized perivascular niches that likely allow asymmetric division of CD8 stem memory T cells to preserve stemness and proliferative capacity while simultaneously generating differentiated cytotoxic CD8 T cells.396 It appears that IL-33 released by perivascular fibroblasts in chronically inflamed tissues balances effector differentiation and stemness of infiltrating CD8 T cells to perpetuate autoimmune tissue destruction or control of viral reactivation and cancer. More broadly, specialized fibroblasts at tissue interfaces (e.g. mucosal/epidermal barriers, blood/parenchyma, tissue/cavity, etc.) may, upon activation, express an array of molecules that contribute to the restimulation or continued activation of infiltrating or activated tissue resident T cells.
If specialized fibroblasts promote local reactivation and differentiation of T cells, can they do the same for B cells? Like what was described for T cells above, Dll1 provided by murine FRC promotes MZ B cell survival and plasma cell differentiation (Figure 4E).291 MZ B cells express both classical (MHC) and non-classical (CD1 family) antigen presentation machinery.291,397–399 MZ-like B cells expressing CD1 family transcripts are found in RA synovial tissue.400 Thus, FRC- or FDC-like fibroblasts in the RA synovium may provide DLL1/Notch2 signals that promote MZ-like B cell survival and plasma cell differentiation in this inflamed tissue. It’s unclear, however, if human FRC or FDC provide similar Dll1/Notch2 signals and, if so, whether this system promotes B cell survival. This may extend to other inflammatory diseases where large B cell infiltrates are common, such as DLE and SS, although this awaits experimental validation.
Follicular dendritic cells (FDC) are specialized fibroblasts found in B cell follicles in SLO. Immune complexes deposit onto FDC, which depends upon complement receptors 1 and 2 (CR1 and CR2) and, in secondary follicles, FcγRIIB expressed by FDC (Figure 4F).215,401–409 Recycling of immune complexes by FDC controls how long B cell responses last in the absence of fresh insults, and how frequently antigen is available to drive affinity maturation of B cells.215,405,410 This function of FDC is important for germinal center reactions and the generation of functional, high-affinity antibodies.213,215,403,406,408,409,411 Immune complex recycling by FDC also means that B cell activation and germinal centers can be maintained long after the initiating insult is cleared.401,402,404–406,410 Do FDC-like fibroblasts perform similar B cell activating functions in tissues affected by IMID?
FDC-like networks are associated with activation-induced cytidine deaminase (AID) expression by accumulating B cells during inflammation in non-lymphoid tissues (Figure 4F).21,159,162 This local AID expression appears to be functional. The presence of secondary follicles in RA synovia, the lungs of RA patients with pulmonary complications and salivary glands of SS patients is associated with higher serum levels of anti-rheumatoid factor. DNA sequencing of B cells from germinal center positive joints suggests local clonal expansion and affinity maturation of autoantibodies.21,200,361,412 Similarly, transplanting RA synovium samples either with or without CD21+ FDC into SCID mice demonstrated that AID expression and mature FDC were associated with local class switching and secretion of human anti-citrullinated protein IgG.210 Murine models of infection and autoimmunity also indicate that B cells can undergo local antigen-specific proliferation and affinity maturation only in the presence of mature FDC (Figure 4F).107,153,154,159,198,204 Synovial levels of an FDC transcriptional signature and individual immune complex trapping genes were positively correlated with both DAS28-ESR and DAS28-CRP supporting the idea that local autoreactive B cell responses are associated with more aggressive or severe disease.303
Local B cell activation by synovial fibroblasts recapitulates in vitro. Co-culture of naïve IgD+ B cells with synovial fibroblasts from RA patients induces AID expression, class switching to and secretion of IgG without any exogenous stimulus. AID expression as well as class switching to and secretion of IgA and IgG is enhanced by TLR3 stimulation during the co-culture.301 TLR3-dependent induction of AID and class switching requires BAFF signalling through its receptors.301 This appeared to occur independent of antigenic stimulation because combined TNFα and LTα1β2 signaling was necessary to increase transcripts of the immune complex trapping genes Complement receptors 1 and 2 (CR1/CD35 and CR2/CD21) and Fragment crystallizable gamma receptor IIb (FCGR2B) by RA synovial fibroblasts.303 The requirement for combined TNFR and LTβR signaling in the full maturation of FDC is supported by in vivo pharmacological blockade and genetic disruption data in mice, rats and monkeys.145,192,194,197,202,413,414 However, there may be other pathways to full FDC maturation as they form in inducible bronchus-associated lymphoid tissue in mice lacking TNFα, LTα and LTα1β2.198
These observations are particularly relevant to developing treatments for patients with autoantibody or B cell-driven autoimmune diseases. First, these local B and plasma cell responses in IMID likely give rise to tissue resident B and plasma cells.208,415–418 Rituximab appears to require B cells to recirculate for clearance via antibody-dependent phagocytosis or complement-dependent cytotoxicity, and thus fails to deplete tissue resident B cells.161,207,419 Other therapeutic approaches including targeting survival factors that affect B cells and/or plasma cells in the bone marrow, SLO and TLS, depleting antibodies that work by antibody-dependent cellular cytotoxicity or chimeric antigen receptor T cell (CAR-T) or combination therapy approaches will likely be necessary for larger clinical benefit in these patients.420–423 Second, because FDC trap and retain antigen for at least 56 days either clearance of immune complexes from FDC or depletion of FDC is necessary to prevent perpetuation or re-emergence of autoreactive B and plasma cell responses.215
Conclusions
Over the past 20 years, increasing interest in the roles that fibroblasts play in both FD and IMID has revolutionized our understanding of these cells. This growing body of work suggests that over the course of FD and IMID, fibroblasts in the affected tissues undergo progressively divergent differentiation which is associated with the distinct clinical outcomes of these diseases. It seems likely that acute injury signals including alarmins, PRR activation and the serum response initiate fibroblast activation early in both FD and IMID. While this has been demonstrated in several animal models of FD and IMID, conclusive proof from patients will require the development of diagnostics that can detect and differentiate these diseases before they manifest clinically. As the types of acute injury signals vary widely, future research into whether these differ between FD and IMID will help us better understand whether differences in fibroblast functions are initiated in the earliest stages of disease.
Currently, it seems that fibroblast activation by alarmins in FD and IMID translates to their recruitment of and growth factor support for monocytes and granulocytes in the early stages of disease. Several therapies targeting the alarmins that activate fibroblasts, among other cells, have entered the clinic or are under development and have met with varied success. IL-1 antagonists have primarily shown efficacy in IMID which have associated familial genetic mutations in pathways involving IL-1. Efficacy in IMID has been mixed, with promising results seen in idiopathic juvenile arthritis and hidradenitis suppurativa, both of which often present with substantial neutrophil infiltrates (reviewed in Ref. 424). The mixed success of IL-1 antagonists may, in part, reflect the redundant stimuli (IL-1, TNFα, serum, etc.) that drive production of neutrophil and monocyte chemokines, growth factors and activation molecules by fibroblasts. Combined blockade of these pathways may be necessary to drive clinical responses in a broader population of patients as was recently proposed.37
After their initial activation by acute injury signals, divergence of fibroblast differentiation states between FD and IMID appears to be a sequential process due, in part, to an evolving dialogue with infiltrating leukocytes. This is supported by the distinct sizes and compositions of leukocyte infiltrates observed when FD and IMID patients present clinically. In IMID where there is a break in tolerance, autoreactive T cells infiltrating the affected tissues retain mature DCs presenting cognate antigen around blood vessels and sites of damage. Clustering of macrophages and DCs with T cells and other tissue resident lymphocytes may trigger subsequent fibroblast differentiation through combinations of TNFR1/2, LTβR and IL-22 signaling along with cytokines produced by type 1, type 2 or type 3 polarized immune cells. Ultimately, this can lead to TLS formation with local priming of autoreactive T cells and class switching, affinity maturation and autoantibody production by B and plasma cells.
The many therapies antagonizing TNFR1/2, IL-13 and IL-17 that have been approved for IMID give physician scientists opportunities to investigate whether any of these signals are required for the maintenance of lymphocyte aggregates or TLSs. As each of these has animal model and in vitro data supporting their direct effects on fibroblast differentiation and lymphocyte aggregate formation some of their efficacy may come from reversing this. Recently, RA pathotypes reflective of distinct fibroblast differentiation have been associated with the course of disease progression and clinical responses to different DMARDs.161,199,303 Enriched efficacy of tocilizumab in RA patients with the diffuse myeloid pathotype fits with the hypothesis that trans-IL-6 signaling between these two cell types is pathogenic in many of these patients.161 This observation also suggests that combined blockade of IL-6 receptor and an orthogonal pathway involved in lymphoid aggregate formation, like TNFR1/2 or LTβR, may increase the breadth of responding patients to those with the lympho-myeloid pathotype as was recently proposed.425
The necessity of combination therapies is further reinforced by the coexistence of multiple fibroblast functional states in the same tissue. This has been best described in RA where fibroblasts specialized for supporting local autoimmune reactivation in lymphoid aggregates and erosive fibroblasts that degrade cartilage occur in the same tissue. Such functionally different fibroblasts arising in the same tissue suggests the existence of distinct microanatomic signaling niches within the tissue. Although targeting different therapeutics to microanatomic niches in the same tissue is beyond our current engineering capabilities, rational target combinations in multispecific large molecules or selectively engineered small molecule polypharmacology may serve as a bridge to the future. Eventually, combining multiple targeted lipid nanoparticle, RNA or small molecule therapies into a single formulation may achieve something resembling microanatomically directed therapies.
Parallel to the observation of these lymphoid niches in IMID is the finding of niches comprised of ECM fibroblasts, Fab5 macrophages and neutrophils in FD. In FD, acute injury activates fibroblasts to produce chemokines that attract neutrophils and monocytes. However, the absence of infiltrating T cells may lead to DC depletion, which allows PDPN+ fibroblasts to contract and stiffen the local ECM. This begins activating latent factors in the ECM, such as TGFβ. Simultaneously, these activated fibroblasts produce GM-CSF to keep infiltrating neutrophils and monocytes alive. Together with neutrophils and γδ T cells producing IL-17A, these activated fibroblasts induce monocytes to differentiate into Fab5 macrophages. ECM fibroblasts and Fab5 macrophages form CDH-11 synapses where the macrophages produce additional latent TGFβ for integrin-mediated activation by fibroblasts. Given the mixed type 2 and type 3 inflammatory tone in some FD it is likely that ECM fibroblasts are also activated by IL-4 and IL-13 produced by ILC2, granulocytes, iNKT and MAIT cells. These type 2 cytokines polarize macrophages away from the Fab5 profile and towards a remodeling state aimed at re-epithelialization and -vascularization.
Like our observation of the need for combination therapies in IMID, the coexistence of mixed inflammatory skewing in FD indicates a similar requirement for combination therapies. Blocking the key type 3 cytokines in some FD appears to drive a compensatory spike in type 2 inflammation. Conversely, inhibiting fibrotic type 2 signaling seems to induce a rebound in type 3 inflammation. Simultaneously inhibiting both systems may be necessary to cover broad FD patient populations. Similarly, as the RA community has begun to define pathotypes associated with distinct clinical outcomes and responses to therapies, FD researchers and clinicians will benefit from more detailed characterization of disease heterogeneity in their patients. Additionally, as the inflammatory phase of FD appears to precede end-stage fibrosis the development of diagnostic methods to identify and classify these patients earlier in their disease progression will likely improve the frequency of successful therapeutic intervention.
Finally, the observation of local, durable immune responses in tissues affected by FD and IMID has consequences for depleting therapies. For example, the lympho-myeloid RA pathotype is the only one with large B cell infiltrates in the affected joints. Why, then, was this pathotype not enriched for rituximab responders?161 Possibly because rituximab was unable to deplete synovial B cells due to a lack of recirculation or low levels of complement-mediated lysis in the joint. Similarly, the niche in FD appears to create a similar challenge for conventional depleting therapies. Three therapeutic strategies currently under investigation may address this challenge.
First, activated cells, whether lymphocytes or fibroblasts, appear to be more susceptible to pharmacological perturbations of the balance between pro- and anti-apoptotic proteins.295,426 Better understanding how these differ between activated fibroblasts in FD and IMID may allow successful small molecule or RNA targeting of these systems. Second, depleting antibodies with Fc regions engineered for efficient antibody-dependent cellular cytotoxicity may circumvent the need for phagocytosis of opsonized cells by Kupffer cells or red pulp macrophages. Third, CAR-T or other engineered cell therapies can allow for targeted depletion of pathogenic cell types throughout the body as recently demonstrated in systemic lupus erythematosus and myasthenia gravis patients, and animal models of cardiac fibrosis.420,427,428 Regardless of the therapeutic approach, we predict that accounting for and disrupting the fibroblasts that lay the foundation for pathogenic niches in FD and IMID will be critical for future therapeutics.
Data availability
No data are associated with this article.
Acknowledgements
The authors thank Thomas A. Wynn, Ken Dower, Richard L. Gieseck III, Matthew V. Russo, Matthew L. Lech, Kevin M. Hart, Katharine L. Horback, Jeffrey L. Browning and Ari B. Molofsky for helpful conversations during preparation of this manuscript.
References
- 1.
Lu CP, Polak L, Keyes BE, et al.:
Spatiotemporal antagonism in mesenchymal-epithelial signaling in sweat versus hair fate decision.
Science.
2016 Dec 23; 354(6319). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 2.
Cox CB, Storm EE, Kapoor VN, et al.:
IL-1R1-dependent signaling coordinates epithelial regeneration in response to intestinal damage.
Sci. Immunol.
2021 May 7; 6(59). PubMed Abstract
| Publisher Full Text
- 3.
Koliaraki V, Chalkidi N, Henriques A, et al.:
Innate Sensing through Mesenchymal TLR4/MyD88 Signals Promotes Spontaneous Intestinal Tumorigenesis.
Cell Rep.
2019 Jan 15; 26(3):536–545.e4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Reyes NS, Krasilnikov M, Allen NC, et al.:
Sentinel p16(INK4a+) cells in the basement membrane form a reparative niche in the lung.
Science.
2022 Oct 14; 378(6616): 192–201. Epub 20221013. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Roulis M, Nikolaou C, Kotsaki E, et al.:
Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis.
Proc. Natl. Acad. Sci. U. S. A.
2014 Oct 28; 111(43): E4658–E4667. Epub 20141014. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Cassandras M, Wang C, Kathiriya J, et al.:
Gli1(+) mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung.
Nat. Cell Biol.
2020 Nov; 22(11): 1295–1306. Epub 20201012. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Filliol A, Saito Y, Nair A, et al.:
Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis.
Nature.
2022 Oct; 610(7931): 356–365. Epub 20221005. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 8.
Jacob JM, Di Carlo SE, Stzepourginski I, et al.:
PDGFRalpha-induced stromal maturation is required to restrain postnatal intestinal epithelial stemness and promote defense mechanisms.
Cell Stem Cell.
2022 May 5; 29(5): 856–868.e5. PubMed Abstract
| Publisher Full Text
- 9.
Benahmed F, Chyou S, Dasoveanu D, et al.:
Multiple CD11c+ cells collaboratively express IL-1beta to modulate stromal vascular endothelial growth factor and lymph node vascular-stromal growth.
J. Immunol.
2014 May 1; 192(9): 4153–4163. Epub 20140321. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Chyou S, Benahmed F, Chen J, et al.:
Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells.
J. Immunol.
2011 Dec 1; 187(11): 5558–5567. Epub 20111026. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Chyou S, Ekland EH, Carpenter AC, et al.:
Fibroblast-type reticular stromal cells regulate the lymph node vasculature.
J. Immunol.
2008 Sep 15; 181(6): 3887–3896. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 12.
Heichler C, Scheibe K, Schmied A, et al.:
STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis.
Gut.
2020 Jul; 69(7): 1269–1282. Epub 20191104. PubMed Abstract
| Publisher Full Text
- 13.
Webster B, Ekland EH, Agle LM, et al.:
Regulation of lymph node vascular growth by dendritic cells.
J. Exp. Med.
2006 Aug 7; 203(8): 1903–1913. Epub 20060710. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
Barron AMS, Mantero JC, Ho JD, et al.:
Perivascular Adventitial Fibroblast Specialization Accompanies T Cell Retention in the Inflamed Human Dermis.
J. Immunol.
2019 Jan 1; 202(1): 56–68. Epub 20181203. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 15.
Brunt EM:
Nonalcoholic steatohepatitis.
Semin. Liver Dis.
2004 Feb; 24(1): 3–20. PubMed Abstract
| Publisher Full Text
- 16.
Kleiner DE, Brunt EM, Van Natta M, et al.:
Design and validation of a histological scoring system for nonalcoholic fatty liver disease.
Hepatology.
2005 Jun; 41(6): 1313–1321. PubMed Abstract
| Publisher Full Text
- 17.
Ramachandran P, Dobie R, Wilson-Kanamori JR, et al.:
Resolving the fibrotic niche of human liver cirrhosis at single-cell level.
Nature.
2019 Nov; 575(7783): 512–518. Epub 20191009. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Fabre T, Barron AMS, Christensen SM, et al.:
Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation.
Sci. Immunol.
2023 Apr 14; 8(82): eadd8945. Epub 20230407. PubMed Abstract
| Publisher Full Text
- 19.
van Batenburg AA , van Oosterhout MFM , Knoppert SN, et al.:
The Extent of Inflammatory Cell Infiltrate and Fibrosis in Lungs of Telomere- and Surfactant-Related Familial Pulmonary Fibrosis.
Front. Med (Lausanne).
2021; 8: 736485. Epub 20210924. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Smith ML:
The histologic diagnosis of usual interstitial pneumonia of idiopathic pulmonary fibrosis. Where we are and where we need to go.
Mod. Pathol.
2022 Jan; 35(Suppl 1): 8–14. Epub 20210831. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Rangel-Moreno J, Hartson L, Navarro C, et al.:
Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis.
J. Clin. Invest.
2006 Dec; 116(12): 3183–3194. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Novak CM, Sethuraman S, Luikart KL, et al.:
Alveolar macrophages drive lung fibroblast function in cocultures of IPF and normal patient samples.
Am. J. Physiol. Lung Cell. Mol. Physiol.
2023 Apr 1; 324(4): L507–L520. Epub 20230215. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Skaug B, Khanna D, Swindell WR, et al.:
Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile.
Ann. Rheum. Dis.
2020 Mar; 79(3): 379–386. Epub 20191125. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Gong T, Liu L, Jiang W, et al.:
DAMP-sensing receptors in sterile inflammation and inflammatory diseases.
Nat. Rev. Immunol.
2020 Feb; 20(2): 95–112. Epub 20190926. PubMed Abstract
| Publisher Full Text
- 25.
Onuh JO, Qiu H:
Serum response factor-cofactor interactions and their implications in disease.
FEBS J.
2021 May; 288(10): 3120–3134. Epub 20200912. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 26.
Yang D, Han Z, Oppenheim JJ:
Alarmins and immunity.
Immunol. Rev.
2017 Nov; 280(1): 41–56. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Cavagnero KJ, Gallo RL:
Essential immune functions of fibroblasts in innate host defense.
Front. Immunol.
2022; 13: 1058862. Epub 20221215. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Zhao S, Dejanovic D, Yao P, et al.:
Selective deletion of MyD88 signaling in alpha-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation.
Mucosal Immunol.
2020 Jul; 13(4): 665–678. Epub 20200204. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Walton KL, Holt L, Sartor RB:
Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways.
Am. J. Physiol. Gastrointest. Liver Physiol.
2009 Mar; 296(3): G601–G611. Epub 20090108. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Proost P, Vynckier AK, Mahieu F, et al.:
Microbial Toll-like receptor ligands differentially regulate CXCL10/IP-10 expression in fibroblasts and mononuclear leukocytes in synergy with IFN-gamma and provide a mechanism for enhanced synovial chemokine levels in septic arthritis.
Eur. J. Immunol.
2003 Nov; 33(11): 3146–3153. PubMed Abstract
| Publisher Full Text
- 31.
Yao C, Oh JH, Lee DH, et al.:
Toll-like receptor family members in skin fibroblasts are functional and have a higher expression compared to skin keratinocytes.
Int. J. Mol. Med.
2015 May; 35(5): 1443–1450. Epub 20150318. PubMed Abstract
| Publisher Full Text
- 32.
Kougias P, Wei D, Rice PJ, et al.:
Normal human fibroblasts express pattern recognition receptors for fungal (1-->3)-beta-D-glucans.
Infect. Immun.
2001 Jun; 69(6): 3933–3938. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Chang HY, Chi JT, Dudoit S, et al.:
Diversity, topographic differentiation, and positional memory in human fibroblasts.
Proc. Natl. Acad. Sci. U. S. A.
2002 Oct 1; 99(20): 12877–12882. Epub 20020924. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Iyer VR, Eisen MB, Ross DT, et al.:
The transcriptional program in the response of human fibroblasts to serum.
Science.
1999 Jan 1; 283(5398): 83–87. PubMed Abstract
| Publisher Full Text
- 35.
Chang HY, Sneddon JB, Alizadeh AA, et al.:
Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds.
PLoS Biol.
2004 Feb; 2(2): E7. Epub 20040113. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Suwara MI, Green NJ, Borthwick LA, et al.:
IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts.
Mucosal Immunol.
2014 May; 7(3): 684–693. Epub 20131030. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 37.
Friedrich M, Pohin M, Jackson MA, et al.:
IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies.
Nat. Med.
2021 Nov; 27(11): 1970–1981. Epub 20211021. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 38.
Scarpa M, Kessler S, Sadler T, et al.:
The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation.
Am. J. Pathol.
2015 Jun; 185(6): 1624–1637. Epub 20150410. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Wolk K, Brembach TC, Simaite D, et al.:
Activity and components of the granulocyte colony-stimulating factor pathway in hidradenitis suppurativa.
Br. J. Dermatol.
2021 Jul; 185(1): 164–176. Epub 20210304. PubMed Abstract
| Publisher Full Text
- 40.
Witte-Handel E, Wolk K, Tsaousi A, et al.:
The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction.
J. Invest. Dermatol.
2019 Jun; 139(6): 1294–1305. Epub 20181205. PubMed Abstract
| Publisher Full Text
- 41.
Elias JA, Reynolds MM, Kotloff RM, et al.:
Fibroblast interleukin 1 beta: synergistic stimulation by recombinant interleukin 1 and tumor necrosis factor and posttranscriptional regulation.
Proc. Natl. Acad. Sci. U. S. A.
1989 Aug; 86(16): 6171–6175. PubMed Abstract
| Free Full Text
- 42.
Elias JA, Reynolds MM:
Interleukin-1 and tumor necrosis factor synergistically stimulate lung fibroblast interleukin-1 alpha production.
Am. J. Respir. Cell Mol. Biol.
1990 Jul; 3(1): 13–20. PubMed Abstract
| Publisher Full Text
- 43.
Elias JA, Gustilo K, Baeder W, et al.:
Synergistic stimulation of fibroblast prostaglandin production by recombinant interleukin 1 and tumor necrosis factor.
J. Immunol.
1987 Jun 1; 138(11): 3812–3816. PubMed Abstract
| Publisher Full Text
- 44.
Paish HL, Kalson NS, Smith GR, et al.:
Fibroblasts Promote Inflammation and Pain via IL-1alpha Induction of the Monocyte Chemoattractant Chemokine (C-C Motif) Ligand 2.
Am. J. Pathol.
2018 Mar; 188(3): 696–714. Epub 20171215. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 45.
Larsen CG, Anderson AO, Oppenheim JJ, et al.:
Production of interleukin-8 by human dermal fibroblasts and keratinocytes in response to interleukin-1 or tumour necrosis factor.
Immunology.
1989 Sep; 68(1): 31–36. PubMed Abstract
| Free Full Text
- 46.
Jeong JG, Kim JM, Cho H, et al.:
Effects of IL-1beta on gene expression in human rheumatoid synovial fibroblasts.
Biochem. Biophys. Res. Commun.
2004 Nov 5; 324(1): 3–7. PubMed Abstract
| Publisher Full Text
- 47.
Bageghni SA, Hemmings KE, Yuldasheva NY:
Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction.
JCI Insight.
2019 Aug 8; 5(17). pii: 125074. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 48.
Yamada T, Fujieda S, Yanagi S, et al.:
IL-1 induced chemokine production through the association of Syk with TNF receptor-associated factor-6 in nasal fibroblast lines.
J. Immunol.
2001 Jul 1; 167(1): 283–288. Epub 20190808. PubMed Abstract
| Publisher Full Text
- 49.
Barlo NP, van Moorsel CH , Korthagen NM, et al.:
Genetic variability in the IL1RN gene and the balance between interleukin (IL)-1 receptor agonist and IL-1beta in idiopathic pulmonary fibrosis.
Clin. Exp. Immunol.
2011 Dec; 166(3): 346–351. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Richter AG, Perkins GD, Chavda A, et al.:
Neutrophil chemotaxis in granulomatosis with polyangiitis (Wegener’s) and idiopathic pulmonary fibrosis.
Eur. Respir. J.
2011 Nov; 38(5): 1081–1088. Epub 20110901. PubMed Abstract
| Publisher Full Text
- 51.
Aumiller V, Balsara N, Wilhelm J, et al.:
WNT/beta-catenin signaling induces IL-1beta expression by alveolar epithelial cells in pulmonary fibrosis.
Am. J. Respir. Cell Mol. Biol.
2013 Jul; 49(1): 96–104. PubMed Abstract
| Publisher Full Text
- 52.
Hussein MR, Hassan HI, Hofny ER, et al.:
Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin 1beta, and tumour necrosis factor alpha in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis.
J. Clin. Pathol.
2005 Feb; 58(2): 178–184. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 53.
Lee JU, Chang HS, Lee HJ, et al.:
Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis.
BMC Pulm. Med.
2017 Feb 15; 17(1): 39. Epub 20170215. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 54.
Maekawa T, Jinnin M, Ohtsuki M, et al.:
Serum levels of interleukin-1alpha in patients with systemic sclerosis.
J. Dermatol.
2013 Feb; 40(2): 98–101. Epub 20121018. PubMed Abstract
| Publisher Full Text
- 55.
Solomonidi N, Vlachoyiannopoulos PG, Pappa M, et al.:
A randomized clinical trial of bermekimab treatment for clinical improvement of systemic sclerosis.
iScience.
2023 Sep 15; 26(9): 107670. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Lettesjo H, Nordstrom E, Strom H, et al.:
Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions.
Scand. J. Immunol.
1998 Sep; 48(3): 286–292. Epub 20230819. PubMed Abstract
| Publisher Full Text
- 57.
Kokebie R, Aggarwal R, Lidder S, et al.:
The role of synovial fluid markers of catabolism and anabolism in osteoarthritis, rheumatoid arthritis and asymptomatic organ donors.
Arthritis Res. Ther.
2011 Mar 24; 13(2): R50. Epub 20110324. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 58.
Hussein MR, Fathi NA, El-Din AM, et al.:
Alterations of the CD4(+), CD8 (+) T cell subsets, interleukins-1beta, IL-10, IL-17, tumor necrosis factor-alpha and soluble intercellular adhesion molecule-1 in rheumatoid arthritis and osteoarthritis: preliminary observations.
Pathol. Oncol. Res.
2008 Sep; 14(3): 321–328. Epub 20080408. PubMed Abstract
| Publisher Full Text
- 59.
Bergman GJ, Hochberg MC, Boers M, et al.:
Indirect comparison of tocilizumab and other biologic agents in patients with rheumatoid arthritis and inadequate response to disease-modifying antirheumatic drugs.
Semin. Arthritis Rheum.
2010 Jun; 39(6): 425–441. Epub 20100311. PubMed Abstract
| Publisher Full Text
- 60.
Cohen SB, Moreland LW, et al.:
A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate.
Ann. Rheum. Dis.
2004 Sep; 63(9): 1062–1068. Epub 20040413. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Xiangyang Z, Lutian Y, Lin Z, et al.:
Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis.
Cytokine.
2012 Apr; 58(1): 6–9. Epub 20120110. PubMed Abstract
| Publisher Full Text
- 62.
Matsuyama Y, Okazaki H, Tamemoto H, et al.:
Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis.
J. Rheumatol.
2010 Jan; 37(1): 18–25. Epub 20091116. PubMed Abstract
| Publisher Full Text
- 63.
Tang S, Huang H, Hu F, et al.:
Increased IL-33 in synovial fluid and paired serum is associated with disease activity and autoantibodies in rheumatoid arthritis.
Clin. Dev. Immunol.
2013; 2013: 985301. Epub 20130909. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Oleszycka E, Moran HB, Tynan GA, et al.:
IL-1alpha and inflammasome-independent IL-1beta promote neutrophil infiltration following alum vaccination.
FEBS J.
2016 Jan; 283(1): 9–24. Epub 20151104. PubMed Abstract
| Publisher Full Text
- 65.
Saxena A, Chen W, Su Y, et al.:
IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium.
J. Immunol.
2013 Nov 1; 191(9): 4838–48. Epub 20130927. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 66.
Bonnardel J, T’Jonck W, Gaublomme D, et al.:
Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche.
Immunity.
2019 Oct 15; 51(4): 638–654.e9. Epub 20190924. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Dahlgren MW, Jones SW, Cautivo KM, et al.:
Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches.
Immunity.
2019 Mar 19; 50(3): 707–722.e6. Epub 20190226. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Aparicio-Domingo P, Cannelle H, Buechler MB, et al.:
Fibroblast-derived IL-33 is dispensable for lymph node homeostasis but critical for CD8 T-cell responses to acute and chronic viral infection.
Eur. J. Immunol.
2021 Jan; 51(1): 76–90. English. PubMed Abstract
| Publisher Full Text
- 69.
Marx AF, Kallert SM, Brunner TM, et al.:
The alarmin interleukin-33 promotes the expansion and preserves the stemness of Tcf-1+CD8+T cells in chronic viral infection.
Immunity.
2023 Apr 11; 56(4): 813–828.e10. English. PubMed Abstract
| Publisher Full Text
- 70.
Malhotra D, Fletcher AL, Astarita J, et al.:
Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks.
Nat. Immunol.
2012 Apr 1; 13(5): 499–510. Epub 20120401. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Dwyer GK, Mathews LR, Villegas JA, et al.:
IL-33 acts as a costimulatory signal to generate alloreactive Th1 cells in graft-versus-host disease.
J. Clin. Invest.
2022 Jun 15; 132(12). English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 72.
Sbierski-Kind J, Cautivo KM, Wagner JC, et al.:
Group 2 innate lymphoid cells constrain type 3/17 lymphocytes in shared stromal niches to restrict liver fibrosis.
bioRxiv.
2023 Apr 28. PubMed Abstract
| Free Full Text
- 73.
Wu J, Li Q, Deng J, et al.:
Association between IL-33 and other inflammatory factors in patients with rheumatoid arthritis and in fibroblast-like synoviocytes in vitro.
Exp. Ther. Med.
2021 Feb; 21(2): 161. Epub 20201218. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Lee EJ, So MW, Hong S, et al.:
Interleukin-33 acts as a transcriptional repressor and extracellular cytokine in fibroblast-like synoviocytes in patients with rheumatoid arthritis.
Cytokine.
2016 Jan; 77: 35–43. Epub 20151029. PubMed Abstract
| Publisher Full Text
- 75.
Kunisch E, Chakilam S, Gandesiri M, et al.:
IL-33 regulates TNF-alpha dependent effects in synovial fibroblasts.
Int. J. Mol. Med.
2012 Apr; 29(4): 530–540. Epub 20120110. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 76.
Savinko T, Matikainen S, Saarialho-Kere U, et al.:
IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors.
J. Invest. Dermatol.
2012 May; 132(5): 1392–1400. Epub 20120126. PubMed Abstract
| Publisher Full Text
- 77.
Cayrol C, Girard JP:
Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine.
Cytokine.
2022 Aug; 156: 155891. Epub 20220525. PubMed Abstract
| Publisher Full Text
- 78.
Spallanzani RG, Zemmour D, Xiao T, et al.:
Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors.
Sci. Immunol.
2019 May 3; 4(35). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Ring SS, Cupovic J, Onder L, et al.:
Viral vector-mediated reprogramming of the fibroblastic tumor stroma sustains curative melanoma treatment.
Nat. Commun.
2021 Aug 5; 12(1): 4734. Epub 20210805. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 80.
Cohen ES, Scott IC, Majithiya JB, et al.:
Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation.
Nat. Commun.
2015 Sep 14; 6: 8327. Epub 20150914. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Hayakawa H, Hayakawa M, Kume A, et al.:
Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation.
J. Biol. Chem.
2007 Sep 7; 282(36): 26369–26380. Epub 20070710. PubMed Abstract
| Publisher Full Text
- 82.
Lefrancais E, Roga S, Gautier V, et al.:
IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G.
Proc. Natl. Acad. Sci. U. S. A.
2012 Jan 31; 109(5): 1673–1678. Epub 20120117. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Capelli A, Di Stefano A, Gnemmi I, et al.:
CCR5 expression and CC chemokine levels in idiopathic pulmonary fibrosis.
Eur. Respir. J.
2005 Apr; 25(4): 701–707. PubMed Abstract
| Publisher Full Text
- 84.
Schmidt K, Martinez-Gamboa L, Meier S, et al.:
Bronchoalveoloar lavage fluid cytokines and chemokines as markers and predictors for the outcome of interstitial lung disease in systemic sclerosis patients.
Arthritis Res. Ther.
2009; 11(4): R111. Epub 20090717. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 85.
Pierer M, Rethage J, Seibl R, et al.:
Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands.
J. Immunol.
2004 Jan 15; 172(2): 1256–1265. PubMed Abstract
| Publisher Full Text
- 86.
Hampel U, Sesselmann S, Iserovich P, et al.:
Chemokine and cytokine levels in osteoarthritis and rheumatoid arthritis synovial fluid.
J. Immunol. Methods.
2013 Oct 31; 396(1-2): 134–139. Epub 20130828. PubMed Abstract
| Publisher Full Text
- 87.
Li L, Jiang BE:
Serum and synovial fluid chemokine ligand 2/monocyte chemoattractant protein 1 concentrations correlates with symptomatic severity in patients with knee osteoarthritis.
Ann. Clin. Biochem.
2015 Mar; 52(Pt 2): 276–282. Epub 20140708. PubMed Abstract
| Publisher Full Text
- 88.
Suga M, Iyonaga K, Ichiyasu H, et al.:
Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases.
Eur. Respir. J.
1999 Aug; 14(2): 376–382. PubMed Abstract
| Publisher Full Text
- 89.
Filer A, Bik M, Parsonage GN, et al.:
Galectin 3 induces a distinctive pattern of cytokine and chemokine production in rheumatoid synovial fibroblasts via selective signaling pathways.
Arthritis Rheum.
2009 Jun; 60(6): 1604–1614. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 90.
Parsonage G, Filer A, Bik M, et al.:
Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha.
Arthritis Res. Ther.
2008; 10(2): R47. Epub 20080423. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 91.
Natsuaki Y, Egawa G, Nakamizo S, et al.:
Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin.
Nat. Immunol.
2014 Nov; 15(11): 1064–1069. Epub 20140921. PubMed Abstract
| Publisher Full Text
- 92.
Ohl L, Mohaupt M, Czeloth N, et al.:
CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions.
Immunity.
2004 Aug; 21(2): 279–288. PubMed Abstract
| Publisher Full Text
- 93.
Saeki H, Moore AM, Brown MJ, et al.:
Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes.
J. Immunol.
1999 Mar 1; 162(5): 2472–2475. PubMed Abstract
| Publisher Full Text
- 94.
Chia JJ, Zhu T, Chyou S, et al.:
Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis.
J. Clin. Invest.
2016 Nov 1; 126(11): 4331–4345. Epub 20161010. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Pradere JP, Kluwe J, De Minicis S, et al.:
Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice.
Hepatology.
2013 Oct; 58(4): 1461–1473. Epub 20130809. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 96.
Goodarzi K, Goodarzi M, Tager AM, et al.:
Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues.
Nat. Immunol.
2003 Oct; 4(10): 965–973. Epub 20030831. PubMed Abstract
| Publisher Full Text
- 97.
Mora JR, Cheng G, Picarella D, et al.:
Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues.
J. Exp. Med.
2005 Jan 17; 201(2): 303–16. Epub 20050110. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 98.
Prizant H, Patil N, Negatu S, et al.:
CXCL10+ peripheral activation niches couple preferred sites of Th1 entry with optimal APC encounter.
Cell Rep.
2021 Aug 10; 36(6): 109523. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 99.
Durrant DM, Daniels BP, Klein RS:
IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile Virus encephalitis.
J. Immunol.
2014 Oct 15; 193(8): 4095–4106. Epub 20140908. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 100.
Durrant DM, Daniels BP, Pasieka T, et al.:
CCR5 limits cortical viral loads during West Nile virus infection of the central nervous system.
J. Neuroinflammation.
2015 Dec 15; 12: 233. Epub 20151215. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 101.
McCandless EE, Zhang B, Diamond MS, et al.:
CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis.
Proc. Natl. Acad. Sci. U. S. A.
2008 Aug 12; 105(32): 11270–11275. Epub 20080804. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 102.
Klein RS, Lin E, Zhang B, et al.:
Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis.
J. Virol.
2005 Sep; 79(17): 11457–11466. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 103.
Sitati E, McCandless EE, Klein RS, et al.:
CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis.
J. Virol.
2007 Sep; 81(18): 9801–9811. Epub 20070711. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 104.
Nazari B, Rice LM, Stifano G, et al.:
Altered Dermal Fibroblasts in Systemic Sclerosis Display Podoplanin and CD90.
Am. J. Pathol.
2016 Oct; 186(10): 2650–2664. Epub 20160823. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 105.
Peduto L, Dulauroy S, Lochner M, et al.:
Inflammation recapitulates the ontogeny of lymphoid stromal cells.
J. Immunol.
2009 May 1; 182(9): 5789–5799. PubMed Abstract
| Publisher Full Text
- 106.
Croft AP, Naylor AJ, Marshall JL, et al.:
Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage.
Arthritis Res. Ther.
2016 Nov 18; 18(1): 270. Epub 20161118. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Nayar S, Campos J, Smith CG, et al.:
Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology.
P. Natl. Acad. Sci. USA.
2019 Jul 2; 116(27): 13490–13497. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 108.
Acton SE, Farrugia AJ, Astarita JL, et al.:
Dendritic cells control fibroblastic reticular network tension and lymph node expansion.
Nature.
2014 Oct 23; 514(7523): 498–502. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Astarita JL, Cremasco V, Fu JX, et al.:
The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture.
Nat. Immunol.
2015 Jan; 16(1): 75–84. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 110.
Astarita JL, Keerthivasan S, Husain B, et al.:
The neutrophil protein CD177 is a novel PDPN receptor that regulates human cancer-associated fibroblast physiology.
PLoS One.
2021; 16(12): e0260800. Epub 20211208. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Sarrazy V, Koehler A, Chow ML, et al.:
Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction.
Cardiovasc. Res.
2014 Jun 1; 102(3): 407–417. Epub 20140317. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 112.
Annes JP, Chen Y, Munger JS, et al.:
Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1.
J. Cell Biol.
2004 Jun 7; 165(5): 723–734. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 113.
Munger JS, Huang X, Kawakatsu H, et al.:
The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis.
Cell.
1999 Feb 5; 96(3): 319–328. PubMed Abstract
| Publisher Full Text
- 114.
Le VQ, Zhao B, Ramesh S, et al.:
A specialized integrin-binding motif enables proTGF-beta2 activation by integrin alphaVbeta6 but not alphaVbeta8.
Proc. Natl. Acad. Sci. U. S. A.
2023 Jun 13; 120(24): e2304874120. Epub 20230606. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 115.
Hart KMFT, Sciurba JS, Gieseck RL III, et al.:
Type-2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β.
Sci. Transl. Med.
2017; 9. In press. PubMed Abstract
| Publisher Full Text
- 116.
Sciurba JC, Gieseck RL, Jiwrajka N, et al.:
Fibroblast-specific integrin-alpha V differentially regulates type 17 and type 2 driven inflammation and fibrosis.
J. Pathol.
2019 May; 248(1): 16–29. Epub 20190124. PubMed Abstract
| Publisher Full Text
- 117.
Decaris ML, Schaub JR, Chen C, et al.:
Dual inhibition of alpha(v)beta(6) and alpha(v)beta(1) reduces fibrogenesis in lung tissue explants from patients with IPF.
Respir. Res.
2021 Oct 19; 22(1): 265. Epub 20211019. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 118.
Seki E, De Minicis S, Osterreicher CH, et al.:
TLR4 enhances TGF-beta signaling and hepatic fibrosis.
Nat. Med.
2007 Nov; 13(11): 1324–32. Epub 20071021. PubMed Abstract
| Publisher Full Text
- 119.
Pakshir P, Alizadehgiashi M, Wong B, et al.:
Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix.
Nat. Commun.
2019 Apr 23; 10(1): 1850. Epub 20190423. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
Plotnikov SV, Pasapera AM, Sabass B, et al.:
Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration.
Cell.
2012 Dec 21; 151(7): 1513–1527. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 121.
Okuma T, Terasaki Y, Kaikita K, et al.:
C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases.
J. Pathol.
2004 Dec; 204(5): 594–604. PubMed Abstract
| Publisher Full Text
- 122.
Venosa A, Cowman S, Katzen J, et al.:
Role of CCR2(+) Myeloid Cells in Inflammation Responses Driven by Expression of a Surfactant Protein-C Mutant in the Alveolar Epithelium.
Front. Immunol.
2021; 12: 665818. Epub 20210422. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Oz HH, Cheng EC, Di Pietro C, et al.:
Recruited monocytes/macrophages drive pulmonary neutrophilic inflammation and irreversible lung tissue remodeling in cystic fibrosis.
Cell Rep.
2022 Dec 13; 41(11): 111797. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Mitchell C, Couton D, Couty JP, et al.:
Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice.
Am. J. Pathol.
2009 May; 174(5): 1766–1775. Epub 20090409. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 125.
Krenkel O, Puengel T, Govaere O, et al.:
Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis.
Hepatology.
2018 Apr67(4): 1270–1283. Epub 20180219. PubMed Abstract
| Publisher Full Text
- 126.
Reuveni D, Gore Y, Leung PSC, et al.:
The Critical Role of Chemokine (C-C Motif) Receptor 2-Positive Monocytes in Autoimmune Cholangitis.
Front. Immunol.
2018; 9: 1852. Epub 20180815. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 127.
Guicciardi ME, Trussoni CE, Krishnan A, et al.:
Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice.
J. Hepatol.
2018 Sep; 69(3): 676–686. Epub 20180524. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 128.
Duffield JS, Forbes SJ, Constandinou CM, et al.:
Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair.
J. Clin. Invest.
2005 Jan; 115(1): 56–65. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 129.
Lodyga M, Cambridge E, Karvonen HM, et al.:
Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β.
Sci. Signal.
2019 Jan 15; 12(564). eng. PubMed Abstract
| Publisher Full Text
- 130.
Jannat RA, Robbins GP, Ricart BG, et al.:
Neutrophil adhesion and chemotaxis depend on substrate mechanics.
J. Phys. Condens. Matter.
2010 May 19; 22(19): 194117. Epub 20190115. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 131.
Oakes PW, Patel DC, Morin NA, et al.:
Neutrophil morphology and migration are affected by substrate elasticity.
Blood.
2009 Aug 13; 114(7): 1387–1395. Epub 20090602. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 132.
He X, Tolosa MF, Zhang T, et al.:
Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis.
JCI Insight.
2022 Feb 22; 7(4). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 133.
Liu F, Lagares D, Choi KM, et al.:
Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis.
Am. J. Physiol. Lung Cell. Mol. Physiol.
2015 Feb 15; 308(4): L344–L357. Epub 20141212. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 134.
Hinz B, Celetta G, Tomasek JJ, et al.:
Alpha-smooth muscle actin expression upregulates fibroblast contractile activity.
Mol. Biol. Cell.
2001 Sep; 12(9): 2730–2741. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 135.
Borensztajn K, Bresser P, van der Loos C , et al.:
Protease-activated receptor-2 induces myofibroblast differentiation and tissue factor up-regulation during bleomycin-induced lung injury: potential role in pulmonary fibrosis.
Am. J. Pathol.
2010 Dec; 177(6): 2753–2764. Epub 20101022. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 136.
Acton SE, Astarita JL, Malhotra D, et al.:
Podoplanin-Rich Stromal Networks Induce Dendritic Cell Motility via Activation of the C-type Lectin Receptor CLEC-2.
Immunity.
2012 Aug 24; 37(2): 276–89. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 137.
Yang CY, Vogt TK, Favre S, et al.:
Trapping of naive lymphocytes triggers rapid growth and remodeling of the fibroblast network in reactive murine lymph nodes.
P. Natl. Acad. Sci. USA.
2014 Jan 7; 111(1): E109–E118. English. PubMed Abstract
| Publisher Full Text
- 138.
Jarjour M, Jorquera A, Mondor I, et al.:
Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells.
J. Exp. Med.
2014 Jun 2; 211(6): 1109–1122. Epub 20140526. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 139.
Assen FP, Abe J, Hons M, et al.:
Multitier mechanics control stromal adaptations in the swelling lymph node.
Nat. Immunol.
2022 Aug; 23(8): 1246–1255. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
Saito Y, Respatika D, Komori S, et al.:
SIRPalpha(+) dendritic cells regulate homeostasis of fibroblastic reticular cells via TNF receptor ligands in the adult spleen.
Proc. Natl. Acad. Sci. U. S. A.
2017 Nov 21; 114(47): E10151–E10160. Epub 20171106. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 141.
Komori S, Saito Y, Respatika D, et al.:
SIRPalpha(+) dendritic cells promote the development of fibroblastic reticular cells in murine peripheral lymph nodes.
Eur. J. Immunol.
2019 Sep; 49(9): 1364–71. Epub 20190603. PubMed Abstract
| Publisher Full Text
- 142.
Kumar V, Dasoveanu DC, Chyou S, et al.:
A dendritic-cell-stromal axis maintains immune responses in lymph nodes.
Immunity.
2015 Apr 21; 42(4): 719–730. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 143.
Suto T, Tosevska A, Dalwigk K, et al.:
TNFR2 is critical for TNF-induced rheumatoid arthritis fibroblast-like synoviocyte inflammation.
Rheumatology (Oxford).
2022 Nov 2; 61(11): 4535–4546. PubMed Abstract
| Publisher Full Text
- 144.
Sakkou M, Chouvardas P, Ntari L, et al.:
Mesenchymal TNFR2 promotes the development of polyarthritis and comorbid heart valve stenosis.
JCI Insight.
Apr 52018; 3(7). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 145.
Ngo VN, Korner H, Gunn MD, et al.:
Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen.
J. Exp. Med.
1999 Jan 18; 189(2): 403–412. Epub 20180405. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 146.
Kocks JR, Davalos-Misslitz AC, Hintzen G, et al.:
Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue.
J. Exp. Med.
2007 Apr 16; 204(4): 723–34. Epub 20070319. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 147.
Fleige H, Bosnjak B, Permanyer M, et al.:
Manifold Roles of CCR7 and Its Ligands in the Induction and Maintenance of Bronchus-Associated Lymphoid Tissue.
Cell Rep.
2018 Apr 17; 23(3): 783–795. PubMed Abstract
| Publisher Full Text
- 148.
Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, et al.:
Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza.
Proc. Natl. Acad. Sci. U. S. A.
2007 Jun 19; 104(25): 10577–82. Epub 20070611. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 149.
Halle S, Dujardin HC, Bakocevic N, et al.:
Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells.
J. Exp. Med.
2009 Nov 23; 206(12): 2593–601. Epub 20091116. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 150.
Fleige H, Ravens S, Moschovakis GL, et al.:
IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs.
J. Exp. Med.
2014 Apr 07211(4): 643–51. Epub 20140324. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 151.
Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M , et al.:
The development of inducible bronchus-associated lymphoid tissue depends on IL-17.
Nat. Immunol.
2011 Jun 12; 12(7): 639–46. Epub 20110612. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 152.
Toyoshima M, Chida K, Sato A:
Antigen uptake and subsequent cell kinetics in bronchus-associated lymphoid tissue.
Respirology.
2000 Jun; 5(2): 141–5. eng. PubMed Abstract
| Publisher Full Text
- 153.
Bombardieri M, Barone F, Lucchesi D, et al.:
Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice.
J. Immunol.
2012 Oct 01; 189(7): 3767–3776. Epub 20120831. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 154.
Barone F, Nayar S, Campos J, et al.:
IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs.
P. Natl. Acad. Sci. USA.
2015 Sep 1; 112(35): 11024–11029. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 155.
Pikor NB, Astarita JL, Summers-Deluca L, et al.:
Integration of Th17-and Lymphotoxin-Derived Signals Initiates Meningeal-Resident Stromal Cell Remodeling to Propagate Neuroinflammation.
Immunity.
2015 Dec 15; 43(6): 1160–1173. English. PubMed Abstract
| Publisher Full Text
- 156.
Barone F, Bombardieri M, Manzo A, et al.:
Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjogren’s syndrome.
Arthritis Rheum.
2005 Jun; 52(6): 1773–1784. PubMed Abstract
| Publisher Full Text
- 157.
Manzo A, Paoletti S, Carulli M, et al.:
Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis.
Eur. J. Immunol.
2005 May; 35(5): 1347–1359. PubMed Abstract
| Publisher Full Text
- 158.
Manzo A, Bugatti S, Caporali R, et al.:
CCL21 expression pattern of human secondary lymphoid organ stroma is conserved in inflammatory lesions with lymphoid neogenesis.
Am. J. Pathol.
2007 Nov; 171(5): 1549–1562. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 159.
Astorri E, Bombardieri M, Gabba S, et al.:
Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets.
J. Immunol.
2010 Sep 15; 185(6): 3359–3368. Epub 20100816. PubMed Abstract
| Publisher Full Text
- 160.
Mittal S, Revell M, Barone F, et al.:
Lymphoid aggregates that resemble tertiary lymphoid organs define a specific pathological subset in metal-on-metal hip replacements.
PLoS One.
2013; 8(5): e63470. Epub 20130528. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 161.
Rivellese F, Surace AEA, Goldmann K, et al.:
Rituximab versus tocilizumab in rheumatoid arthritis: synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial.
Nat. Med.
2022 Jun; 28(6): 1256–1268. Epub 20220519. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 162.
Bombardieri M, Barone F, Humby F, et al.:
Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjogren’s syndrome.
J. Immunol.
2007 Oct 1; 179(7): 4929–4938. PubMed Abstract
| Publisher Full Text
- 163.
Wang C, Hyams B, Allen NC, et al.:
Dysregulated lung stroma drives emphysema exacerbation by potentiating resident lymphocytes to suppress an epithelial stem cell reservoir.
Immunity.
2023 Mar 14; 56(3): 576–591.e10. Epub 20230222. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 164.
Yanni G, Whelan A, Feighery C, et al.:
Analysis of cell populations in rheumatoid arthritis synovial tissues.
Semin. Arthritis Rheum.
1992 Jun; 21(6): 393–399. PubMed Abstract
| Publisher Full Text
- 165.
Lowin T, Anssar TM, Bauml M, et al.:
Positive and negative cooperativity of TNF and Interferon-gamma in regulating synovial fibroblast function and B cell survival in fibroblast/B cell co-cultures.
Sci. Rep.
2020 Jan 21; 10(1): 780. Epub 20200121. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 166.
Antonelli A, Ferrari SM, Fallahi P, et al.:
Monokine induced by interferon gamma (IFNgamma) (CXCL9) and IFNgamma inducible T-cell alpha-chemoattractant (CXCL11) involvement in Graves’ disease and ophthalmopathy: modulation by peroxisome proliferator-activated receptor-gamma agonists.
J. Clin. Endocrinol. Metab.
2009 May; 94(5): 1803–1809. Epub 20090310. eng. PubMed Abstract
| Publisher Full Text
- 167.
Yan R, Moresco P, Gegenhuber B, et al.:
T cell-Mediated Development of Stromal Fibroblasts with an Immune-Enhancing Chemokine Profile.
Cancer Immunol. Res.
2023 Jun 07: OF1–OF16. Epub 20230607. eng. PubMed Abstract
| Publisher Full Text
- 168.
Ohmori Y, Schreiber RD, Hamilton TA:
Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB.
J. Biol. Chem.
1997 Jun 6; 272(23): 14899–14907. PubMed Abstract
| Publisher Full Text
- 169.
Hiroi M, Ohmori Y:
The transcriptional coactivator CREB-binding protein cooperates with STAT1 and NF-kappa B for synergistic transcriptional activation of the CXC ligand 9/monokine induced by interferon-gamma gene.
J. Biol. Chem.
2003 Jan 3; 278(1): 651–60. Epub 20021025. PubMed Abstract
| Publisher Full Text
- 170.
Korsunsky I, Millard N, Fan J, et al.:
Fast, sensitive and accurate integration of single-cell data with Harmony.
Nat. Methods.
2019 Dec; 16(12): 1289–1296. Epub 2019/11/20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 171.
Zhang F, Wei K, Slowikowski K, et al.:
Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry.
Nat. Immunol.
2019 Jul; 20(7): 928–942. Epub 20190506. PubMed Abstract
| Free Full Text
- 172.
Cremasco V, Astarita JL, Grauel AL, et al.:
FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors.
Cancer Immunol. Res.
2018 Dec; 6(12): 1472–1485. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 173.
Dominguez CX, Muller S, Keerthivasan S, et al.:
Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy.
Cancer Discov.
2020 Feb; 10(2): 232–253. Epub 20191107. PubMed Abstract
| Publisher Full Text
- 174.
Fletcher AL, Malhotra D, Acton SE, et al.:
Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells.
Front. Immunol.
2011; 2: 35. Epub 20110912. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 175.
Steinmetz OM, Turner JE, Paust HJ, et al. et al.:
CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis.
J. Immunol.
2009 Oct 1; 183(7): 4693–4704. Epub 20090904. PubMed Abstract
| Publisher Full Text
- 176.
Oo YH, Banz V, Kavanagh D, et al.:
CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver.
J. Hepatol.
2012 Nov; 57(5): 1044–1051. Epub 20120714. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 177.
Qu Y, Wen J, Thomas G, et al.:
Baseline Frequency of Inflammatory Cxcl9-Expressing Tumor-Associated Macrophages Predicts Response to Avelumab Treatment.
Cell Rep.
2020 Jul 7; 32(1): 107873. PubMed Abstract
| Publisher Full Text
- 178.
Coursey TG, Gandhi NB, Volpe EA, et al.:
Chemokine receptors CCR6 and CXCR3 are necessary for CD4(+) T cell mediated ocular surface disease in experimental dry eye disease.
PLoS One.
2013; 8(11): e78508. Epub 20131104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 179.
Slowikowski K, Nguyen HN, Noss EH, et al.:
CUX1 and IκBζ (NFKBIZ) mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts.
Proc. Natl. Acad. Sci. U. S. A.
2020 Mar 10; 117(10): 5532–5541. Epub 20200220. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 180.
Wu L, Ong S, Talor MV, et al.:
Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy.
J. Exp. Med.
2014 Jun 30; 211(7): 1449–1464. Epub 20140616. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 181.
Kouri VP, Olkkonen J, Nurmi K, et al.:
IL-17A and TNF synergistically drive expression of proinflammatory mediators in synovial fibroblasts via IκBζ-dependent induction of ELF3.
Rheumatology (Oxford).
2023 Feb 01; 62(2): 872–885. eng. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 182.
Hirota K, Yoshitomi H, Hashimoto M, et al.:
Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model.
J. Exp. Med.
2007 Nov 26; 204(12): 2803–2812. Epub 20071119. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 183.
Luther SA, Tang HL, Hyman PL, et al.:
Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse.
P. Natl. Acad. Sci. USA.
2000 Nov 7; 97(23): 12694–9. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Chai Q, Onder L, Scandella E, et al.:
Maturation of Lymph Node Fibroblastic Reticular Cells from Myofibroblastic Precursors Is Critical for Antiviral Immunity.
Immunity.
2013 May 23; 38(5): 1013–1024. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 185.
Link A, Vogt TK, Favre S, et al.:
Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells.
Nat. Immunol.
2007 Nov; 8(11): 1255–1265. Epub 20070923. PubMed Abstract
| Publisher Full Text
- 186.
Stein JV, Rot A, Luo Y, et al.:
The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules.
J. Exp. Med.
2000 Jan 3; 191(1): 61–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 187.
Baekkevold ES, Yamanaka T, Palframan RT, et al.:
The CCR7 ligand elc (CCL19) is transcytosed in high endothelial venules and mediates T cell recruitment.
J. Exp. Med.
2001 May 7; 193(9): 1105–1112. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 188.
Link A, Hardie DL, Favre S, et al.:
Association of T-Zone Reticular Networks and Conduits with Ectopic Lymphoid Tissues in Mice and Humans.
Am. J. Pathol.
2011 Apr; 178(4): 1662–1675. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 189.
Korsunsky I, Wei K, Pohin M, et al.:
Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases.
Med-Cambridge.
2022 Jul 8; 3(7): 481–518.e14. English. PubMed Abstract
| Publisher Full Text
- 190.
Buechler MB, Pradhan RN, Krishnamurty AT, et al.:
Cross-tissue organization of the fibroblast lineage.
Nature.
2021 May; 593(7860): 575–579. Epub 20210512. PubMed Abstract
| Publisher Full Text
- 191.
Kugler M, Dellinger M, Kartnig F, et al.:
Cytokine-directed cellular cross-talk imprints synovial pathotypes in rheumatoid arthritis.
Ann. Rheum. Dis.
2023 Jun 21; 82: ard-2022-223396–ard-2022-221152. Epub 20230621.PubMed Abstract
| Publisher Full Text
- 192.
Seleznik GM, Reding T, Romrig F, et al.:
Lymphotoxin beta receptor signaling promotes development of autoimmune pancreatitis.
Gastroenterology.
2012 Nov; 143(5): 1361–1374. Epub 20120802. PubMed Abstract
| Publisher Full Text
- 193.
Zhang Z, Ma X, Bai J, et al.:
Characterize the lavage and serum cytokine profiles of interstitial pneumonia with autoimmune features and their implications for progressive fibrosis.
Rheumatology (Oxford).
2023 Aug 22. Epub 20230822. PubMed Abstract
| Publisher Full Text
- 194.
Fava RA, Kennedy SM, Wood SG, et al.:
Lymphotoxin-beta receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjogren’s syndrome.
Arthritis Res. Ther.
2011; 13(6): R182. Epub 20111101. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 195.
Schaeuble K, Britschgi MR, Scarpellino L, et al.:
Perivascular Fibroblasts of the Developing Spleen Act as LTalpha1beta2-Dependent Precursors of Both T and B Zone Organizer Cells.
Cell Rep.
2017 Nov 28; 21(9): 2500–2514. PubMed Abstract
- 196.
Nayar S, Pontarini E, Campos J, et al.:
Immunofibroblasts regulate LTalpha3 expression in tertiary lymphoid structures in a pathway dependent on ICOS/ICOSL interaction.
Commun. Biol.
2022 May 4; 5(1): 413. Epub 20220504. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 197.
Gatumu MK, Skarstein K, Papandile A, et al.:
Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjogren’s syndrome in salivary glands of non-obese diabetic mice.
Arthritis Res. Ther.
2009; 11(1): R24. Epub 20090218. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 198.
Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, et al.:
Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity.
Nat. Med.
2004 Sep; 10(9): 927–934. Epub 20040815. PubMed Abstract
| Publisher Full Text
- 199.
Humby F, Lewis M, Ramamoorthi N, et al.:
Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients.
Ann. Rheum. Dis.
2019 Jun; 78(6): 761–72. Epub 20190316. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 200.
Randen I, Mellbye OJ, Forre O, et al.:
The identification of germinal centres and follicular dendritic cell networks in rheumatoid synovial tissue.
Scand. J. Immunol.
1995 May; 41(5): 481–486. PubMed Abstract
| Publisher Full Text
- 201.
Aziz KE, McCluskey PJ, Wakefield D:
Characterisation of follicular dendritic cells in labial salivary glands of patients with primary Sjogren syndrome: comparison with tonsillar lymphoid follicles.
Ann. Rheum. Dis.
1997 Feb; 56(2): 140–143. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 202.
Ansel KM, Ngo VN, Hyman PL, et al.:
A chemokine-driven positive feedback loop organizes lymphoid follicles.
Nature.
2000 Jul 20; 406(6793): 309–314. English. PubMed Abstract
| Publisher Full Text
- 203.
Amft N, Curnow SJ, Scheel-Toellner D, et al.:
Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren’s syndrome.
Arthritis Rheum.
2001 Nov; 44(11): 2633–2641. PubMed Abstract
| Publisher Full Text
- 204.
Luther SA, Lopez T, Bai W, et al.:
BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis.
Immunity.
2000 May; 12(5): 471–481. English. PubMed Abstract
| Publisher Full Text
- 205.
Benezech C, Luu NT, Walker JA, et al.:
Inflammation-induced formation of fat-associated lymphoid clusters.
Nat. Immunol.
2015 Aug; 16(8): 819–828. Epub 20150629. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 206.
Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, et al.:
Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens.
Immunity.
2009 May; 30(5): 731–743. Epub 20090507. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 207.
Barker KA, Etesami NS, Shenoy AT, et al.:
Lung-resident memory B cells protect against bacterial pneumonia.
J. Clin. Invest.
2021 Jun 1; 131(11). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 208.
Allie SR, Bradley JE, Mudunuru U, et al.:
The establishment of resident memory B cells in the lung requires local antigen encounter.
Nat. Immunol.
2019 Jan; 20(1): 97–108. Epub 20181203. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 209.
Argyriou A, Wadsworth MH 2nd, Lendvai A, et al.:
Single cell sequencing identifies clonally expanded synovial CD4(+) T (PH) cells expressing GPR56 in rheumatoid arthritis.
Nat. Commun.
2022 Jul 13; 13(1): 4046. Epub 20220713. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 210.
Humby F, Bombardieri M, Manzo A, et al.:
Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium.
PLoS Med.
2009 Jan 13; 6(1): e1. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 211.
Dubey LK, Ludewig B, Luther SA, et al.:
IL-4Ralpha-Expressing B Cells Are Required for CXCL13 Production by Fibroblastic Reticular Cells.
Cell Rep.
2019 May 21; 27(8): 2442–2458.e5. PubMed Abstract
| Publisher Full Text
- 212.
van de Pavert SA , Olivier BJ, Goverse G, et al.:
Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation.
Nat. Immunol.
2009 Nov; 10(11): 1193–9. Epub 20090927. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 213.
Wang X, Cho B, Suzuki K, et al.:
Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers.
J. Exp. Med.
2011 Nov 21; 208(12): 2497–2510. Epub 20111031. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 214.
Khader SA, Guglani L, Rangel-Moreno J, et al.:
IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung.
J. Immunol.
2011 Nov 15; 187(10): 5402–7. Epub 20111014. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 215.
Martinez-Riano A, Wang S, Boeing S, et al.:
Long-term retention of antigens in germinal centers is controlled by the spatial organization of the follicular dendritic cell network.
Nat. Immunol.
2023 Aug; 24(8): 1281–1294. Epub 20230713. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 216.
Buechler MB, Fu W, et al.:
Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer.
Immunity.
2021 May 11; 54(5): 903–915. PubMed Abstract
| Publisher Full Text
- 217.
Adler M, Mayo A, Zhou X, et al.:
Endocytosis as a stabilizing mechanism for tissue homeostasis.
Proc. Natl. Acad. Sci. U. S. A.
2018 Feb 20; 115(8): E1926–E1935. Epub 20180202. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 218.
Zhou X, Franklin RA, Adler M, et al.:
Circuit Design Features of a Stable Two-Cell System.
Cell.
2018 Feb 8; 172(4): 744–757.e17. Epub 20180201. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 219.
Wang S, Li K, Pickholz E, et al.:
An autocrine signaling circuit in hepatic stellate cells underlies advanced fibrosis in nonalcoholic steatohepatitis.
Sci. Transl. Med.
2023 Jan 4; 15(677): eadd3949. Epub 20230104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 220.
Bell AL, Magill MK, McKane WR, et al.:
Measurement of colony-stimulating factors in synovial fluid: potential clinical value.
Rheumatol. Int.
1995; 14(5): 177–182. PubMed Abstract
| Publisher Full Text
- 221.
Meehan RT, Regan EA, Hoffman ED, et al.:
Synovial Fluid Cytokines, Chemokines and MMP Levels in Osteoarthritis Patients with Knee Pain Display a Profile Similar to Many Rheumatoid Arthritis Patients.
J. Clin. Med.
2021 Oct 28; 10(21). Epub 20211028. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 222.
Darrieutort-Laffite C, Boutet MA, Chatelais M, et al.:
IL-1beta and TNFalpha promote monocyte viability through the induction of GM-CSF expression by rheumatoid arthritis synovial fibroblasts.
Mediat. Inflamm.
2014; 2014: 241840. Epub 20141117. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 223.
Stanley S, Mok CC, Vanarsa K, et al.:
Identification of Low-Abundance Urinary Biomarkers in Lupus Nephritis Using Electrochemiluminescence Immunoassays.
Arthritis Rheumatol.
2019 May; 71(5): 744–755. Epub 20190403. PubMed Abstract
| Publisher Full Text
- 224.
Martin-Nares E, Hernandez-Molina G, Lima G, et al.:
Tear levels of IL-7, IL-1alpha, and IL-1beta may differentiate between IgG4-related disease and Sjogren’s syndrome.
Clin. Rheumatol.
2023 Apr; 42(4): 1101–1105. Epub 20230111. PubMed Abstract
| Publisher Full Text
- 225.
Carreno E, Enriquez-de-Salamanca A, Teson M, et al.:
Cytokine and chemokine levels in tears from healthy subjects.
Acta Ophthalmol.
2010 Nov; 88(7): e250–e258. Epub 20100825. PubMed Abstract
| Publisher Full Text
- 226.
Chen X, Aqrawi LA, Utheim TP, et al.:
Elevated cytokine levels in tears and saliva of patients with primary Sjogren’s syndrome correlate with clinical ocular and oral manifestations.
Sci. Rep.
2019 May 13; 9(1): 7319. Epub 20190513. PubMed Abstract
| Free Full Text
- 227.
Taniguchi H, Katoh S, Kadota J, et al.:
Interleukin 5 and granulocyte-macrophage colony-stimulating factor levels in bronchoalveolar lavage fluid in interstitial lung disease.
Eur. Respir. J.
2000 Nov; 16(5): 959–964. PubMed Abstract
| Publisher Full Text
- 228.
Ashitani J, Mukae H, Taniguchi H, et al.:
Granulocyte-colony stimulating factor levels in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis.
Thorax.
1999 Nov; 54(11): 1015–1020. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 229.
Tan-Garcia A, Lai F, Sheng Yeong JP, et al.:
Liver fibrosis and CD206(+) macrophage accumulation are suppressed by anti-GM-CSF therapy.
JHEP Rep.
2020 Feb; 2(1): 100062. Epub 20191206. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 230.
Emad A, Emad Y:
Increased granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) levels in BAL fluid from patients with sulfur mustard gas-induced pulmonary fibrosis.
J. Aerosol Med.
2007 Fall; 20(3): 352–360. PubMed Abstract
| Publisher Full Text
- 231.
Bellomo A, Gentek R, Golub R, et al.:
Macrophage-fibroblast circuits in the spleen.
Immunol. Rev.
2021 Jul; 302(1): 104–125. Epub 20210524. PubMed Abstract
| Publisher Full Text
- 232.
D’Rozario J, Knoblich K, Lutge M, et al.:
Fibroblastic reticular cells provide a supportive niche for lymph node-resident macrophages.
Eur. J. Immunol.
2023 Mar 29; e2250355. Epub 20230329. PubMed Abstract
- 233.
Rodda LB, Lu E, Bennett ML, et al.:
Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity.
Immunity.
2018 May 15; 48(5): 1014–1028.e6. Epub 20180508. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 234.
Alexandre YO, Schienstock D, Lee HJ, et al.:
A diverse fibroblastic stromal cell landscape in the spleen directs tissue homeostasis and immunity.
Sci Immunol.
2022 Jan 7; 7(67): eabj0641. Epub 20220107. PubMed Abstract
| Publisher Full Text
- 235.
Bellomo A, Mondor I, Spinelli L, et al.:
Reticular Fibroblasts Expressing the Transcription Factor WT1 Define a Stromal Niche that Maintains and Replenishes Splenic Red Pulp Macrophages.
Immunity.
2020 Jul 14; 53(1): 127–142.e7. Epub 20200619. PubMed Abstract
| Publisher Full Text
- 236.
Buechler MB, Kim KW, Onufer EJ, et al.:
A Stromal Niche Defined by Expression of the Transcription Factor WT1 Mediates Programming and Homeostasis of Cavity-Resident Macrophages.
Immunity.
2019 Jul 16; 51(1): 119–130.e5. Epub 20190620. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 237.
Schneider C, Nobs SP, Kurrer M, et al.:
Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages.
Nat. Immunol.
2014 Nov; 15(11): 1026–1037. Epub 20140928. PubMed Abstract
| Publisher Full Text
- 238.
Suzuki T, Arumugam P, Sakagami T, et al.:
Pulmonary macrophage transplantation therapy.
Nature.
2014 Oct 23; 514(7523): 450–454. Epub 20141001. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 239.
van de Laar L , Saelens W, De Prijck S, et al.:
Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages.
Immunity.
2016 Apr 19; 44(4): 755–768. Epub 20160315. PubMed Abstract
| Publisher Full Text
- 240.
Guilliams M, De Kleer I, Henri S, et al.:
Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF.
J. Exp. Med.
2013 Sep 23; 210(10): 1977–1992. Epub 20130916. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 241.
Zsengeller ZK, Reed JA, Bachurski CJ, et al.:
Adenovirus-mediated granulocyte-macrophage colony-stimulating factor improves lung pathology of pulmonary alveolar proteinosis in granulocyte-macrophage colony-stimulating factor-deficient mice.
Hum. Gene Ther.
1998 Sep 20; 9(14): 2101–2109. PubMed Abstract
| Publisher Full Text
- 242.
Reed JA, Ikegami M, Cianciolo ER, et al.:
Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice.
Am. J. Phys.
1999 Apr; 276(4): L556–L563. PubMed Abstract
| Publisher Full Text
- 243.
Huffman JA, Hull WM, Dranoff G, et al.:
Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice.
J. Clin. Invest.
1996 Feb 1; 97(3): 649–655. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 244.
Suzuki T, Sakagami T, Rubin BK, et al.:
Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA.
J. Exp. Med.
2008 Nov 24; 205(12): 2703–2710. Epub 20081027. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 245.
Gschwend J, Sherman SPM, Ridder F, et al.:
Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth.
J. Exp. Med.
2021 Oct 4; 218(10). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 246.
Fuentelsaz-Romero S, Cuervo A, Estrada-Capetillo L, et al.:
GM-CSF Expression and Macrophage Polarization in Joints of Undifferentiated Arthritis Patients Evolving to Rheumatoid Arthritis or Psoriatic Arthritis.
Front. Immunol.
2020; 11: 613975. Epub 20210825. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 247.
Hirota K, Hashimoto M, Ito Y, et al.:
Autoimmune Th17 Cells Induced Synovial Stromal and Innate Lymphoid Cell Secretion of the Cytokine GM-CSF to Initiate and Augment Autoimmune Arthritis.
Immunity.
2018 Jun 19; 48(6): 1220–1232.e5. Epub 20180522. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 248.
Louis C, Souza-Fonseca-Guimaraes F, Yang Y, et al.:
NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS.
J. Exp. Med.
2020 May 4; 217(5). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 249.
Boettcher S, Gerosa RC, Radpour R, et al.:
Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis.
Blood.
2014 Aug 28; 124(9): 1393–1403. Epub 20140702. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 250.
Falkenburg JH, Harrington MA, Walsh WK, et al.:
Gene-expression and release of macrophage-colony stimulating factor in quiescent and proliferating fibroblasts. Effects of serum, fibroblast growth-promoting factors, and IL-1.
J. Immunol.
1990 Jun 15; 144(12): 4657–4662. PubMed Abstract
| Publisher Full Text
- 251.
Tobler A, Meier R, Seitz M, et al.:
Glucocorticoids downregulate gene expression of GM-CSF, NAP-1/IL-8, and IL-6, but not of M-CSF in human fibroblasts.
Blood.
1992 Jan 1; 79(1): 45–51. PubMed Abstract
| Publisher Full Text
- 252.
Filer A, Parsonage G, Smith E, et al.:
Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils.
Arthritis Rheum.
2006 Jul; 54(7): 2096–2108. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 253.
Cautivo KM, Matatia PR, Lizama CO, et al.:
Interferon gamma constrains type 2 lymphocyte niche boundaries during mixed inflammation.
Immunity.
2022 Feb 8; 55(2): 254–271.e7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 254.
Fabre T, Molina MF, Soucy G, et al.:
Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis.
Sci. Immunol.
2018 Oct 26; 3(28). Epub 2018/10/28. PubMed Abstract
| Publisher Full Text
- 255.
Frangogiannis N:
Transforming growth factor-beta in tissue fibrosis.
J. Exp. Med.
2020 Mar 2; 217(3): e20190103. Epub 2018. Epub 20200213. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 256.
Lodyga M, Hinz B:
TGF-beta1 - A truly transforming growth factor in fibrosis and immunity.
Semin. Cell Dev. Biol.
2020 May; 101: 123–39. Epub 20191224. PubMed Abstract
| Publisher Full Text
- 257.
Batlle E, Massague J:
Transforming Growth Factor-beta Signaling in Immunity and Cancer.
Immunity.
2019 Apr 16; 50(4): 924–940. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 258.
Chen W:
TGF-beta Regulation of T Cells.
Annu. Rev. Immunol.
2023 Apr 26; 41: 483–512. Epub 20230207. PubMed Abstract
| Publisher Full Text
- 259.
Danielpour D, Kim KY, Dart LL, et al.:
Sandwich enzyme-linked immunosorbent assays (SELISAs) quantitate and distinguish two forms of transforming growth factor-beta (TGF-beta 1 and TGF-beta 2) in complex biological fluids.
Growth Factors.
1989; 2(1): 61–71. PubMed Abstract
| Publisher Full Text
- 260.
Taylor AW:
Review of the activation of TGF-beta in immunity.
J. Leukoc. Biol.
2009 Jan; 85(1): 29–33. Epub 20080925. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 261.
van Roon JA , Verweij MC, Wijk MW, et al.:
Increased intraarticular interleukin-7 in rheumatoid arthritis patients stimulates cell contact-dependent activation of CD4(+) T cells and macrophages.
Arthritis Rheum.
2005 Jun; 52(6): 1700–1710. PubMed Abstract
| Publisher Full Text
- 262.
Harada S, Yamamura M, Okamoto H, et al.:
Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis.
Arthritis Rheum.
1999 Jul; 42(7): 1508–1516. PubMed Abstract
| Publisher Full Text
- 263.
McInnes IB, al-Mughales J, Field M, et al.:
The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis.
Nat. Med.
1996 Feb; 2(2): 175–182. PubMed Abstract
| Publisher Full Text
- 264.
Santos Savio A, Machado Diaz AC, Chico Capote A, et al.:
Differential expression of pro-inflammatory cytokines IL-15Ralpha, IL-15, IL-6 and TNFalpha in synovial fluid from rheumatoid arthritis patients.
BMC Musculoskelet. Disord.
2015 Mar 12; 16: 51. Epub 20150312. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 265.
Scanzello CR, Umoh E, Pessler F, et al.:
Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease.
Osteoarthr. Cartil.
2009 Aug; 17(8): 1040–1048. Epub 20090306. PubMed Abstract
| Publisher Full Text
- 266.
Pickens SR, Chamberlain ND, Volin MV, et al.:
Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis.
Arthritis Rheum.
2011 Oct; 63(10): 2884–2893. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 267.
Shinoda K, Hirahara K, Iinuma T, et al.:
Thy1+IL-7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation.
Proc. Natl. Acad. Sci. U. S. A.
2016 May 17; 113(20): E2842–E2851. Epub 20160502. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 268.
Sheikh A, Jackson J, Shim HB, et al.:
Selective dependence on IL-7 for antigen-specific CD8 T cell responses during airway influenza infection.
Sci. Rep.
2022 Jan 7; 12(1): 135. Epub 20220107. PubMed Abstract
| Free Full Text
- 269.
Cui G, Hara T, Simmons S, et al.:
Characterization of the IL-15 niche in primary and secondary lymphoid organs in vivo.
Proc. Natl. Acad. Sci. U. S. A.
2014 Feb 4; 111(5): 1915–1920. Epub 20140121. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 270.
Mortier E, Woo T, Advincula R, et al.:
IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation.
J. Exp. Med.
2008 May 12; 205(5): 1213–25. Epub 20080505. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 271.
Mortier E, Advincula R, Kim L, et al.:
Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets.
Immunity.
2009 Nov 20; 31(5): 811–22. Epub 20091112. PubMed Abstract
| Publisher Full Text
- 272.
Sandau MM, Schluns KS, Lefrancois L, et al.:
Cutting edge: transpresentation of IL-15 by bone marrow-derived cells necessitates expression of IL-15 and IL-15R alpha by the same cells.
J. Immunol.
2004 Dec 1; 173(11): 6537–6541. PubMed Abstract
| Publisher Full Text
- 273.
Perez-Shibayama C, Gil-Cruz C, Cheng HW, et al.:
Fibroblastic reticular cells initiate immune responses in visceral adipose tissues and secure peritoneal immunity.
Sci. Immunol.
2018 Aug; 3(26). English. PubMed Abstract
| Publisher Full Text
- 274.
Becklund BR, Purton JF, Ramsey C, et al.:
The aged lymphoid tissue environment fails to support naive T cell homeostasis.
Sci. Rep.
2016 Aug 2; 6: 30842. Epub 20160802. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 275.
Onder L, Narang P, Scandella E, et al.:
IL-7-producing stromal cells are critical for lymph node remodeling.
Blood.
2012 Dec 6; 120(24): 4675–4683. Epub 20120906. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 276.
Hara T, Shitara S, Imai K, et al.:
Identification of IL-7-producing cells in primary and secondary lymphoid organs using IL-7-GFP knock-in mice.
J. Immunol.
2012 Aug 15; 189(4): 1577–1584. Epub 20120711. PubMed Abstract
| Publisher Full Text
- 277.
Zehentmeier S, Lim VY, Ma Y, et al.:
Dysregulated stem cell niches and altered lymphocyte recirculation cause B and T cell lymphopenia in WHIM syndrome.
Sci. Immunol.
2022 Sep 23; 7(75): eabo3170. Epub 20220923. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 278.
Yang J, Cornelissen F, Papazian N, et al.:
IL-7-dependent maintenance of ILC3s is required for normal entry of lymphocytes into lymph nodes.
J. Exp. Med.
2018 Apr 2; 215(4): 1069–1077. Epub 20180222. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 279.
Knop L, Deiser K, Bank U, et al.:
IL-7 derived from lymph node fibroblastic reticular cells is dispensable for naive T cell homeostasis but crucial for central memory T cell survival.
Eur. J. Immunol.
2020 Jun; 50(6): 846–857. Epub 20200316. PubMed Abstract
| Publisher Full Text
- 280.
Terashima A, Okamoto K, Nakashima T, et al.:
Sepsis-Induced Osteoblast Ablation Causes Immunodeficiency.
Immunity.
2016 Jun 21; 44(6): 1434–1443. Epub 20160614. PubMed Abstract
| Publisher Full Text
- 281.
Chennupati V, Koch U, Coutaz M, et al.:
Notch Signaling Regulates the Homeostasis of Tissue-Restricted Innate-like T Cells.
J. Immunol.
2016 Aug 1; 197(3): 771–782. English. PubMed Abstract
| Publisher Full Text
- 282.
Frank-Bertoncelj M, Trenkmann M, Klein K, et al.:
Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions.
Nat. Commun.
2017 Mar 23; 8: 14852. Epub 20170323. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 283.
von Freeden-Jeffry U , Vieira P, Lucian LA, et al.:
Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine.
J. Exp. Med.
1995 Apr 1; 181(4): 1519–1526. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 284.
Croft AP, Campos J, Jansen K, et al.:
Distinct fibroblast subsets drive inflammation and damage in arthritis.
Nature.
2019 Jun 13; 570(7760): 246–251. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 285.
Pacheco Y, Sole V, Billaud E, et al.:
Despite an impaired response to IL-7, CD4+EM T cells from HIV-positive patients proliferate normally in response to IL-15 and its superagonist, RLI.
AIDS.
2011 Sep 10; 25(14): 1701–1710. PubMed Abstract
| Publisher Full Text
- 286.
Ota N, Takase M, Uchiyama H, et al.:
No requirement of trans presentations of IL-15 for human CD8 T cell proliferation.
J. Immunol.
2010 Nov 15; 185(10): 6041–6048. Epub 20101006. PubMed Abstract
| Publisher Full Text
- 287.
Cremasco V, Woodruff MC, Onder L, et al.:
B cell homeostasis and follicle confines are governed by fibroblastic reticular cells.
Nat. Immunol.
2014 Oct; 15(10): 973–981. Epub 20140824. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 288.
Dubey LK, Lebon L, Mosconi I, et al.:
Lymphotoxin-Dependent B Cell-FRC Crosstalk Promotes De Novo Follicle Formation and Antibody Production following Intestinal Helminth Infection.
Cell Rep.
2016 May 17; 15(7): 1527–1541. English. PubMed Abstract
| Publisher Full Text
- 289.
Gorelik L, Gilbride K, Dobles M, et al.:
Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells.
J. Exp. Med.
2003 Sep 15; 198(6): 937–945. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 290.
Fasnacht N, Huang HY, Koch U, et al.:
Specific fibroblastic niches in secondary lymphoid organs orchestrate distinct Notch-regulated immune responses.
J. Exp. Med.
2014 Oct 20; 211(11): 2265–79. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 291.
Gomez Atria D, Gaudette BT, Londregan J, et al.:
Stromal Notch ligands foster lymphopenia-driven functional plasticity and homeostatic proliferation of naive B cells.
J. Clin. Invest.
2022 Jul 1; 132(13). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 292.
Hase H, Kanno Y, Kojima M, et al.:
BAFF/BLyS can potentiate B-cell selection with the B-cell coreceptor complex.
Blood.
2004 Mar 15; 103(6): 2257–2265. Epub 20031120. PubMed Abstract
| Publisher Full Text
- 293.
Huang HY, Rivas-Caicedo A, Renevey F, et al.:
Identification of a new subset of lymph node stromal cells involved in regulating plasma cell homeostasis.
P. Natl. Acad. Sci. USA.
2018 Jul 17; 115(29): E6826–E6835. English. PubMed Abstract
| Publisher Full Text
- 294.
Korsunsky I, Wei K, Pohin M, et al.:
Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases.
Med-Cambridge.
2022 Jul 8; 3(7): 481–518.e14. Epub 20220531. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 295.
Lesley R, Xu Y, Kalled SL, et al.:
Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF.
Immunity.
2004 Apr; 20(4): 441–453. PubMed Abstract
| Publisher Full Text
- 296.
Craxton A, Magaletti D, Ryan EJ, et al.:
Macrophage- and dendritic cell--dependent regulation of human B-cell proliferation requires the TNF family ligand BAFF.
Blood.
2003 Jun 1; 101(11): 4464–4471. Epub 20030116. PubMed Abstract
| Publisher Full Text
- 297.
Kim HA, Seo GY, Kim PH:
Macrophage-derived BAFF induces AID expression through the p38MAPK/CREB and JNK/AP-1 pathways.
J. Leukoc. Biol.
2011 Mar; 89(3): 393–398. Epub 20101217. PubMed Abstract
| Publisher Full Text
- 298.
Sabat R, Simaite D, Gudjonsson JE, et al.:
Neutrophilic granulocyte-derived B-cell activating factor supports B cells in skin lesions in hidradenitis suppurativa.
J. Allergy Clin. Immunol.
2023 Apr; 151(4): 1015–1026. Epub 20221205. PubMed Abstract
| Publisher Full Text
- 299.
Parsa R, Lund H, Georgoudaki AM, et al.:
BAFF-secreting neutrophils drive plasma cell responses during emergency granulopoiesis.
J. Exp. Med.
2016 Jul 25; 213(8): 1537–1553. Epub 20160718. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 300.
Giordano D, Kuley R, Draves KE, et al.:
BAFF Produced by Neutrophils and Dendritic Cells Is Regulated Differently and Has Distinct Roles in Antibody Responses and Protective Immunity against West Nile Virus.
J. Immunol.
2020 Mar 15; 204(6): 1508–1520. Epub 20200207. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 301.
Bombardieri M, Kam NW, Brentano F, et al.:
A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells.
Ann. Rheum. Dis.
2011 Oct; 70(10): 1857–1865. Epub 20110727. PubMed Abstract
| Publisher Full Text
- 302.
Burger JA, Zvaifler NJ, Tsukada N, et al.:
Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism.
J. Clin. Invest.
2001 Feb; 107(3): 305–315. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 303.
El Shikh MEM, El Sayed R, Aly NAR, et al.:
Follicular dendritic cell differentiation is associated with distinct synovial pathotype signatures in rheumatoid arthritis.
Front Med (Lausanne).
2022; 9: 1013660. Epub 20221116. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 304.
Younesi FS, Son DO, Firmino J, et al.:
Myofibroblast Markers and Microscopy Detection Methods in Cell Culture and Histology.
Methods Mol. Biol.
2021; 2299: 17–47. PubMed Abstract
| Publisher Full Text
- 305.
Ho JD, Chung HJ, Ms Barron A, et al.:
Extensive CD34-to-CD90 Fibroblast Transition Defines Regions of Cutaneous Reparative, Hypertrophic, and Keloidal Scarring.
Am. J. Dermatopathol.
2019 Jan; 41(1): 16–28. eng. PubMed Abstract
| Publisher Full Text
- 306.
Choy DF, Hart KM, Borthwick LA, et al.:
TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma.
Sci. Transl. Med.
2015 Aug 19; 7(301): 301ra129. Epub 2015/08/21. eng. PubMed Abstract
| Publisher Full Text
- 307.
Wilson MS, Wynn TA:
Pulmonary fibrosis: pathogenesis, etiology and regulation.
Mucosal Immunol.
2009 Mar; 2(2): 103–21. Epub 20090107. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 308.
Gieseck RL 3rd, Wilson MS, Wynn TA:
Type 2 immunity in tissue repair and fibrosis.
Nat. Rev. Immunol.
2017 Aug 30. Epub 2017/08/31. PubMed Abstract
- 309.
Wilson MS, Madala SK, Ramalingam TR, et al.:
Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent.
J. Exp. Med.
2010 Mar 15; 207(3): 535–552. Epub 20100222. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 310.
Hams E, Armstrong ME, Barlow JL, et al.:
IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis.
Proc. Natl. Acad. Sci. U. S. A.
2014 Jan 7; 111(1): 367–372. Epub 20131216. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 311.
Landi A, Weismuller TJ, Lankisch TO, et al.:
Differential serum levels of eosinophilic eotaxins in primary sclerosing cholangitis, primary biliary cirrhosis, and autoimmune hepatitis.
J. Interf. Cytokine Res.
2014 Mar; 34(3): 204–214. Epub 20131029. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 312.
Murray LA, Argentieri RL, Farrell FX, et al.:
Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2.
Int. J. Biochem. Cell Biol.
2008; 40(10): 2174–2182. Epub 20080223. PubMed Abstract
| Publisher Full Text
- 313.
Jakubzick C, Choi ES, Carpenter KJ, et al.:
Human pulmonary fibroblasts exhibit altered interleukin-4 and interleukin-13 receptor subunit expression in idiopathic interstitial pneumonia.
Am. J. Pathol.
2004 Jun; 164(6): 1989–2001. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 314.
Sugimoto R, Enjoji M, Nakamuta M, et al.:
Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells.
Liver Int.
2005 Apr; 25(2): 420–428. PubMed Abstract
| Publisher Full Text
- 315.
Gieseck RL 3rd, Ramalingam TR, Hart KM, et al.:
Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis.
Immunity.
2016 Jul 19; 45(1): 145–58. Epub 20160712. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 316.
Kaviratne M, Hesse M, Leusink M, et al.:
IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent.
J. Immunol.
2004 Sep 15; 173(6): 4020–4029. PubMed Abstract
| Publisher Full Text
- 317.
Zhu Z, Homer RJ, Wang Z, et al.:
Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production.
J. Clin. Invest.
1999 Mar; 103(6): 779–788. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 318.
Lee CG, Homer RJ, Zhu Z, et al.:
Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1).
J. Exp. Med.
2001 Sep 17; 194(6): 809–822. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 319.
Cescon M, Gattazzo F, Chen P, et al.:
Collagen VI at a glance.
J. Cell Sci.
2015 Oct 1; 128(19): 3525–3531. Epub 20150916. PubMed Abstract
| Publisher Full Text
- 320.
Mashiko S, Bouguermouh S, Rubio M, et al.:
Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis.
J. Allergy Clin. Immunol.
2015 Aug; 136(2): 351–359.e1. Epub 20150316. PubMed Abstract
| Publisher Full Text
- 321.
Lin AM, Rubin CJ, Khandpur R, et al.:
Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis.
J. Immunol.
2011 Jul 1; 187(1): 490–500. Epub 20110523. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 322.
Noordenbos T, Blijdorp I, Chen S, et al.:
Human mast cells capture, store, and release bioactive, exogenous IL-17A.
J. Leukoc. Biol.
2016 Sep; 100(3): 453–462. Epub 20160331. PubMed Abstract
| Publisher Full Text
- 323.
Meng F, Wang K, Aoyama T, et al.:
Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice.
Gastroenterology.
2012 Sep; 143(3): 765–776.e3. Epub 20120608. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 324.
Kleinschmidt D, Giannou AD, McGee HM, et al.:
A Protective Function of IL-22BP in Ischemia Reperfusion and Acetaminophen-Induced Liver Injury.
J. Immunol.
2017 Dec 15; 199(12): 4078–4090. Epub 20171106. PubMed Abstract
| Publisher Full Text
- 325.
Rolla S, Alchera E, Imarisio C, et al.:
The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice.
Clin. Sci. (Lond.).
2016 Feb; 130(3): 193–203. Epub 20151111. PubMed Abstract
| Publisher Full Text
- 326.
Xue J, Zhao Q, Sharma V, et al.:
Aryl Hydrocarbon Receptor Ligands in Cigarette Smoke Induce Production of Interleukin-22 to Promote Pancreatic Fibrosis in Models of Chronic Pancreatitis.
Gastroenterology.
2016 Dec; 151(6): 1206–1217. Epub 20161018. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 327.
Yao Z, Fanslow WC, Seldin MF, et al.:
Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor.
Immunity.
1995 Dec; 3(6): 811–821. PubMed Abstract
| Publisher Full Text
- 328.
McGeachy MJ, Cua DJ, Gaffen SL:
The IL-17 Family of Cytokines in Health and Disease.
Immunity.
2019 Apr 16; 50(4): 892–906. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 329.
Fabre T, Kared H, Friedman SL, et al.:
IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner.
J. Immunol.
2014 Oct 15; 193(8): 3925–3933. Epub 20140910. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 330.
Pickert G, Neufert C, Leppkes M, et al.:
STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing.
J. Exp. Med.
2009 Jul 6; 206(7): 1465–1472. Epub 20090629. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 331.
Takahashi J, Yamamoto M, Yasukawa H, et al.:
Interleukin-22 Directly Activates Myocardial STAT3 (Signal Transducer and Activator of Transcription-3) Signaling Pathway and Prevents Myocardial Ischemia Reperfusion Injury.
J. Am. Heart Assoc.
2020 Apr 21; 9(8): e014814. Epub 20200417. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 332.
Saxton RA, Henneberg LT, Calafiore M, et al.:
The tissue protective functions of interleukin-22 can be decoupled from pro-inflammatory actions through structure-based design.
Immunity.
2021 Apr 13; 54(4): 660–672.e9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 333.
Pavlidis P, Tsakmaki A, Pantazi E, et al.:
Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy.
Nat. Commun.
2022 Oct 3; 13(1): 5820. Epub 20221003. PubMed Abstract
| Free Full Text
- 334.
Mizoguchi F, Slowikowski K, Wei K, et al.:
Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis.
Nat. Commun.
2018 Feb 23; 9: 789. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 335.
Micheroli R, Elhai M, Edalat S, et al.:
Role of synovial fibroblast subsets across synovial pathotypes in rheumatoid arthritis: a deconvolution analysis.
RMD Open.
2022 Jan; 8(1): e001949. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 336.
Moran EM, Mullan R, McCormick J, et al.:
Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies.
Arthritis Res. Ther.
2009; 11(4): R113. Epub 20090723. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 337.
Pretzel D, Pohlers D, Weinert S, et al.:
In vitro model for the analysis of synovial fibroblast-mediated degradation of intact cartilage.
Arthritis Res. Ther.
2009; 11(1): R25. Epub 20090218. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 338.
Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, et al.:
The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases.
Int. J. Mol. Sci.
2020 Dec 20; 21(24). Epub 20201220. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 339.
Ricard-Blum S:
The collagen family.
Cold Spring Harb. Perspect. Biol.
2011 Jan 1; 3(1): a004978. Epub 20110101. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 340.
Wells JM, Gaggar A, Blalock JE:
MMP generated matrikines.
Matrix Biol.
2015 May-Jul; 44-46: 122–129. Epub 20150128. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 341.
Seidman JS, Troutman TD, Sakai M, et al.:
Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis.
Immunity.
2020 Jun 16; 52(6): 1057–1074.e7. Epub 2020/05/05. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 342.
Xiong X, Kuang H, Ansari S, et al.:
Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis.
Mol. Cell.
2019 Aug 8; 75(3): 644–660.e5. Epub 2019 /08/10. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 343.
Remmerie A, Martens L, Thone T, et al.:
Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver.
Immunity.
2020 Sep 15; 53(3): 641–657.e14. Epub 2020/09/06. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 344.
Joshi N, Watanabe S, Verma R, et al.:
A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages.
Eur. Respir. J.
2020 Jan; 55(1): 1900646. Epub 2019 /10/12. R. Verma has nothing to disclose. Conflict of interest: R.P. Jablonski has nothing to disclose. Conflict of interest: C-I Chen has nothing to disclose. Conflict of interest: P. Cheresh has nothing to disclose. Conflict of interest: N.S. Markov has nothing to disclose. Conflict of interest: P.A. Reyfman has nothing to disclose. Conflict of interest: A.C. McQuattie-Pimentel has nothing to disclose. Conflict of interest: L. Sichizya has nothing to disclose. Conflict of interest: Z. Lu has nothing to disclose. Conflict of interest: R. Piseaux-Aillon has nothing to disclose. Conflict of interest: D. Kirchenbuechler has nothing to disclose. Conflict of interest: A.S. Flozak has nothing to disclose. Conflict of interest: C.J. Gottardi has nothing to disclose. Conflict of interest: C.M. Cuda has nothing to disclose. Conflict of interest: H. Perlman has nothing to disclose. Conflict of interest: M. Jain has nothing to disclose. Conflict of interest: D.W. Kamp has nothing to disclose. Conflict of interest: G.R.S. Budinger has nothing to disclose. Conflict of interest: A.V. Misharin has nothing to disclose. Conflict of interest: N. Joshi has nothing to disclose. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 345.
Reyfman PA, Walter JM, Joshi N, et al.:
Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis.
Am. J. Respir. Crit. Care Med.
2019 Jun 15; 199(12): 1517–1536. Epub 2018/12/18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 346.
Adams TS, Schupp JC, Poli S, et al.:
Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis.
Sci. Adv.
2020 Jul; 6(28): eaba1983. Epub 2020/08/25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 347.
Morse C, Tabib T, Sembrat J, et al.:
Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis.
Eur. Respir. J.
2019 Aug; 54(2): 1802441. Epub 2019/06/22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 348.
Hou J, Zhang J, Cui P, et al.:
TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis.
J. Clin. Invest.
2021 Feb 15; 131(4). Epub 2021/02/16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 349.
Daemen S, Gainullina A, Kalugotla G, et al.:
Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH.
Cell Rep.
2021 Jan 12; 34(2): 108626. Epub 2021/01/14. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 350.
Jaitin DA, Adlung L, Thaiss CA, et al.:
Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner.
Cell.
2019 Jul 25; 178(3): 686–698.e14. Epub 2019/07/02. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 351.
Gao X, Jia G, Guttman A, et al.:
Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis.
Cell Rep Med.
2020 Nov 17; 1(8): 100140. Epub 20201117. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 352.
Minutti CM, Modak RV, Macdonald F, et al.:
A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation.
Immunity.
2019 Mar 19; 50(3): 645–654.e6. Epub 20190212. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 353.
Camara A, Cordeiro OG, Alloush F, et al.:
Lymph Node Mesenchymal and Endothelial Stromal Cells Cooperate via the RANK-RANKL Cytokine Axis to Shape the Sinusoidal Macrophage Niche.
Immunity.
2019 Jun 18; 50(6): 1467–1481.e6. Epub 20190611. PubMed Abstract
| Publisher Full Text
- 354.
Camara A, Lavanant AC, Abe J, et al.:
CD169(+) macrophages in lymph node and spleen critically depend on dual RANK and LTbetaR signaling.
Proc. Natl. Acad. Sci. U. S. A.
2022 Jan 18; 119(3). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 355.
Habbeddine M, Verthuy C, Rastoin O, et al.:
Receptor Activator of NF-kappaB Orchestrates Activation of Antiviral Memory CD8 T Cells in the Spleen Marginal Zone.
Cell Rep.
2017 Nov 28; 21(9): 2515–2527. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 356.
Rose T, Szelinski F, Lisney A, et al.:
SIGLEC1 is a biomarker of disease activity and indicates extraglandular manifestation in primary Sjogren’s syndrome.
RMD Open.
2016; 2(2): e000292. Epub 20161230. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 357.
Lerkvaleekul B, Veldkamp SR, van der Wal MM , et al.:
Siglec-1 expression on monocytes is associated with the interferon signature in juvenile dermatomyositis and can predict treatment response.
Rheumatology (Oxford).
2022 May 5; 61(5): 2144–2155. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 358.
Chou CH, Jain V, Gibson J, et al.:
Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis.
Sci. Rep.
2020 Jul 2; 10(1): 10868. Epub 20200702. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 359.
Jonsson AH, Zhang F, Dunlap G, et al.:
Granzyme K(+) CD8 T cells form a core population in inflamed human tissue.
Sci. Transl. Med.
2022 Jun 15; 14(649): eabo0686. Epub 20220615. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 360.
Wang R, Singaraju A, Marks KE, et al.:
Clonally expanded CD38(hi) cytotoxic CD8 T cells define the T cell infiltrate in checkpoint inhibitor-associated arthritis.
Sci. Immunol.
2023 Jul 28; 8(85): eadd1591. Epub 20230728. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 361.
Dunlap G, Wagner A, Meednu N, et al.:
Clonal associations of lymphocyte subsets and functional states revealed by single cell antigen receptor profiling of T and B cells in rheumatoid arthritis synovium.
bioRxiv.
2023 Mar 21. Epub 20230321. PubMed Abstract
| Free Full Text
- 362.
Weinand K, Sakaue S, Nathan A, et al.:
The Chromatin Landscape of Pathogenic Transcriptional Cell States in Rheumatoid Arthritis. bioRxiv.
2023 Apr 8. Epub 20230408. PubMed Abstract
| Free Full Text
- 363.
Inamo J, Keegan J, Griffith A, et al.:
Deep immunophenotyping reveals circulating activated lymphocytes in individuals at risk for rheumatoid arthritis.
bioRxiv.
2023 Jul 4. Epub 20230704. PubMed Abstract
| Free Full Text
- 364.
Xiong J, Onal M, Jilka RL, et al.:
Matrix-embedded cells control osteoclast formation.
Nat. Med.
2011 Sep 11; 17(10): 1235–41. Epub 20110911. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 365.
Hu Y, Li X, Zhi X, et al.:
RANKL from bone marrow adipose lineage cells promotes osteoclast formation and bone loss.
EMBO Rep.
2021 Jul 5; 22(7): e52481. Epub 20210613. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 366.
Danks L, Komatsu N, Guerrini MM, et al.:
RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation.
Ann. Rheum. Dis.
2016 Jun; 75(6): 1187–95. Epub 20150529. PubMed Abstract
| Publisher Full Text
- 367.
Buckley L, Humphrey MB:
Glucocorticoid-Induced Osteoporosis.
N. Engl. J. Med.
2018 Dec 27; 379(26): 2547–2556. PubMed Abstract
| Publisher Full Text
- 368.
Carli L, Tani C, Spera V, et al.:
Risk factors for osteoporosis and fragility fractures in patients with systemic lupus erythematosus.
Lupus Sci Med.
2016; 3(1): e000098. Epub 20160119. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 369.
Kim S, Yamazaki M, Zella LA, et al.:
Activation of receptor activator of NF-kappaB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers.
Mol Cell Biol.
2006 Sep; 26(17): 6469–6486. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 370.
Quinn JM, Horwood NJ, Elliott J, et al.:
Fibroblastic stromal cells express receptor activator of NF-kappa B ligand and support osteoclast differentiation.
J. Bone Miner. Res.
2000 Aug; 15(8): 1459–1466. PubMed Abstract
| Publisher Full Text
- 371.
Witt JD, Swann M:
Metal wear and tissue response in failed titanium alloy total hip replacements.
J. Bone Joint Surg. Br.
1991 Jul; 73-B(4): 559–563. PubMed Abstract
| Publisher Full Text
- 372.
McGrath LR, Shardlow DL, Ingham E, et al.:
A retrieval study of capital hip prostheses with titanium alloy femoral stems.
J. Bone Joint Surg. Br.
2001 Nov; 83-B(8): 1195–1201. PubMed Abstract
| Publisher Full Text
- 373.
Haynes DR, Crotti TN, Potter AE, et al.:
The osteoclastogenic molecules RANKL and RANK are associated with periprosthetic osteolysis.
J. Bone Joint Surg. Br.
2001 Aug; 83-B(6): 902–911. PubMed Abstract
| Publisher Full Text
- 374.
Wei X, Zhang X, Zuscik MJ, et al.:
Fibroblasts express RANKL and support osteoclastogenesis in a COX-2-dependent manner after stimulation with titanium particles.
J. Bone Miner. Res.
2005 Jul; 20(7): 1136–1148. Epub 20050214. PubMed Abstract
| Publisher Full Text
- 375.
Hashizume M, Hayakawa N, Mihara M:
IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17.
Rheumatology (Oxford).
2008 Nov; 47(11): 1635–1640. Epub 20080911. PubMed Abstract
| Publisher Full Text
- 376.
Lambert ME, Ronai ZA, Weinstein IB, et al.:
Enhancement of major histocompatibility class I protein synthesis by DNA damage in cultured human fibroblasts and keratinocytes.
Mol Cell Biol.
1989 Feb; 9(2): 847–850. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 377.
Ng CT, Nayak BP, Schmedt C, et al.:
Immortalized clones of fibroblastic reticular cells activate virus-specific T cells during virus infection.
Proc. Natl. Acad. Sci. U. S. A.
2012 May 15; 109(20): 7823–7828. Epub 20120501. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 378.
Douglas MW, Kesson AM, King NJ:
CTL recognition of west Nile virus-infected fibroblasts is cell cycle dependent and is associated with virus-induced increases in class I MHC antigen expression.
Immunology.
1994 Aug; 82(4): 561–570. PubMed Abstract
| Free Full Text
- 379.
Jones D, Blackmon A, Neff CP, et al.:
Varicella-Zoster Virus Downregulates Programmed Death Ligand 1 and Major Histocompatibility Complex Class I in Human Brain Vascular Adventitial Fibroblasts, Perineurial Cells, and Lung Fibroblasts.
J. Virol.
2016 Dec 1; 90(23): 10527–10534. Epub 20161114. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 380.
Butz EA, Bevan MJ:
Differential presentation of the same MHC class I epitopes by fibroblasts and dendritic cells.
J. Immunol.
1998 Mar 1; 160(5): 2139–2144. PubMed Abstract
| Publisher Full Text
- 381.
Cheng Y, King NJ, Kesson AM:
Major histocompatibility complex class I (MHC-I) induction by West Nile virus: involvement of 2 signaling pathways in MHC-I up-regulation.
J. Infect. Dis.
2004 Feb 15; 189(4): 658–668. Epub 20040205. PubMed Abstract
| Publisher Full Text
- 382.
Abendroth A, Lin I, Slobedman B, et al.:
Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells.
J. Virol.
2001 May; 75(10): 4878–4888. PubMed Abstract
| Free Full Text
- 383.
Kerdidani D, Aerakis E, Verrou KM, et al.:
Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts.
J. Exp. Med.
2022 Feb 7; 219(2). Epub 20220114. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 384.
Elyada E, Bolisetty M, Laise P, et al.:
Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts.
Cancer Discov.
2019 Aug; 9(8): 1102–1123. Epub 20190613. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 385.
Mueller SN, Vanguri VK, Ha SJ, et al.:
PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice.
J. Clin. Invest.
2010 Jul; 120(7): 2508–2515. Epub 20100614. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 386.
Knoblich K, Cruz Migoni S, Siew SM, et al.:
The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation.
PLoS Biol.
2018 Sep; 16(9): e2005046. Epub 20180904. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 387.
Lukacs-Kornek V, Malhotra D, Fletcher AL, et al.:
Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes.
Nat. Immunol.
2011 Nov; 12(11): 1096–1104. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 388.
Lee JW, Epardaud M, Sun J, et al.:
Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self.
Nat. Immunol.
2007 Feb; 8(2): 181–190. English. PubMed Abstract
- 389.
Schaeuble K, Cannelle H, Favre S, et al.:
Attenuation of chronic antiviral T-cell responses through constitutive COX2-dependent prostanoid synthesis by lymph node fibroblasts.
PLoS Biol.
2019 Jul; 17(7): e3000072. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 390.
Siegert S, Huang HY, Yang CY, et al.:
Fibroblastic Reticular Cells From Lymph Nodes Attenuate T Cell Expansion by Producing Nitric Oxide.
PLoS One.
2011 Nov 14; 6(11): e27618. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 391.
Chung J, Ebens CL, Perkey E, et al.:
Fibroblastic niches prime T cell alloimmunity through Delta-like Notch ligands.
J. Clin. Invest.
2017 Apr 3; 127(4): 1574–1588. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 392.
Tkachev V, Vanderbeck A, Perkey E, et al.:
Notch signaling drives intestinal graft-versus-host disease in mice and nonhuman primates.
Sci. Transl. Med.
2023 Jun 28; 15(702): eadd1175. Epub 20230628. PubMed Abstract
| Publisher Full Text
- 393.
Clark RA, Yamanaka K, Bai M, et al.:
Human skin cells support thymus-independent T cell development.
J. Clin. Invest.
2005 Nov; 115(11): 3239–3249. Epub 20051013. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 394.
Auderset F, Schuster S, Fasnacht N, et al.:
Notch signaling regulates follicular helper T cell differentiation.
J. Immunol.
2013 Sep 1; 191(5): 2344–2350. Epub 20130805. PubMed Abstract
| Publisher Full Text
- 395.
Corria-Osorio J, Carmona SJ, Stefanidis E, et al.:
Orthogonal cytokine engineering enables novel synthetic effector states escaping canonical exhaustion in tumor-rejecting CD8(+) T cells.
Nat. Immunol.
2023 Apr 20; 24: 869–883. English. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 396.
Hua Y, Vella G, Rambow F, et al.:
Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1(+) T lymphocyte niches through a feed-forward loop.
Cancer Cell.
2022 Dec 12; 40(12): 1600–1618.e10. Epub 20221123. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 397.
Weller S, Braun MC, Tan BK, et al.:
Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire.
Blood.
2004 Dec 1; 104(12): 3647–3654. Epub 20040610. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 398.
Amano M, Baumgarth N, Dick MD, et al.:
CD1 expression defines subsets of follicular and marginal zone B cells in the spleen: beta 2-microglobulin-dependent and independent forms.
J. Immunol.
1998 Aug 15; 161(4): 1710–1717. PubMed Abstract
| Publisher Full Text
- 399.
Makowska A, Faizunnessa NN, Anderson P, et al.:
CD1high B cells: a population of mixed origin.
Eur. J. Immunol.
1999 Oct; 29(10): 3285–3294. PubMed Abstract
| Publisher Full Text
- 400.
Palm AK, Friedrich HC, Mezger A, et al.:
Function and regulation of self-reactive marginal zone B cells in autoimmune arthritis.
Cell. Mol. Immunol.
2015 Jul; 12(4): 493–504. Epub 20150511. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 401.
Mandel TE, Phipps RP, Abbot AP, et al.:
Long-term antigen retention by dendritic cells in the popliteal lymph node of immunized mice.
Immunology.
1981 Jun; 43(2): 353–362. PubMed Abstract
| Free Full Text
- 402.
Cirelli KM, Carnathan DG, Nogal B, et al.:
Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance.
Cell.
2019 May 16; 177(5): 1153–1171.e28. Epub 20190509. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 403.
Gordon L, Mabbott N, Wells J, et al.:
Foot-and-mouth disease virus localisation on follicular dendritic cells and sustained induction of neutralising antibodies is dependent on binding to complement receptors (CR2/CR1).
PLoS Pathog.
2022 May; 18(5): e1009942. Epub 20220505. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 404.
Heesters BA, Lindqvist M, Vagefi PA, et al.:
Follicular Dendritic Cells Retain Infectious HIV in Cycling Endosomes.
PLoS Pathog.
2015 Dec; 11(12): e1005285. Epub 20151201. PubMed Abstract
| Free Full Text
- 405.
Heesters BA, Chatterjee P, Kim YA, et al.:
Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation.
Immunity.
2013 Jun 27; 38(6): 1164–1175. Epub 20130613. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 406.
Qin D, Wu J, Vora KA, et al.:
Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response.
J. Immunol.
2000 Jun 15; 164(12): 6268–6275. PubMed Abstract
| Publisher Full Text
- 407.
Yoshida K, van den Berg TK , Dijkstra CD:
Two functionally different follicular dendritic cells in secondary lymphoid follicles of mouse spleen, as revealed by CR1/2 and FcR gamma II-mediated immune-complex trapping.
Immunology.
1993 Sep; 80(1): 34–39. PubMed Abstract
| Free Full Text
- 408.
Fang Y, Xu C, Fu YX, et al.:
Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response.
J. Immunol.
1998 Jun 1; 160(11): 5273–5279. PubMed Abstract
| Publisher Full Text
- 409.
Zhang YN, Lazarovits J, Poon W, et al.:
Nanoparticle Size Influences Antigen Retention and Presentation in Lymph Node Follicles for Humoral Immunity.
Nano Lett.
2019 Oct 9; 19(10): 7226–7235. Epub 20190917. PubMed Abstract
| Publisher Full Text
- 410.
Arulraj T, Binder SC, Meyer-Hermann M:
Rate of Immune Complex Cycling in Follicular Dendritic Cells Determines the Extent of Protecting Antigen Integrity and Availability to Germinal Center B Cells.
J. Immunol.
2021 Apr 1; 206(7): 1436–1442. Epub 20210219. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 411.
Rana M, La Bella A, Lederman R, et al.:
Follicular dendritic cell dysfunction contributes to impaired antigen-specific humoral responses in sepsis-surviving mice.
J. Clin. Invest.
2021 Jun 15; 131(12). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 412.
Broeren MGA, Wang JJ, Balzaretti G, et al.:
Proteogenomic analysis of the autoreactive B cell repertoire in blood and tissues of patients with Sjogren’s syndrome.
Ann. Rheum. Dis.
2022 May; 81(5): 644–652. Epub 20220210. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 413.
Fava RA, Notidis E, Hunt J, et al.:
A role for the lymphotoxin/LIGHT axis in the pathogenesis of murine collagen-induced arthritis.
J. Immunol.
2003 Jul 1; 171(1): 115–126. PubMed Abstract
| Publisher Full Text
- 414.
Gommerman JL, Mackay F, Donskoy E, et al.:
Manipulation of lymphoid microenvironments in nonhuman primates by an inhibitor of the lymphotoxin pathway.
J. Clin. Invest.
2002 Nov; 110(9): 1359–1369. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 415.
MacLean AJ, Richmond N, Koneva L, et al.:
Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites.
Immunity.
2022 Apr 12; 55(4): 718–733.e8. Epub 20220328. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 416.
Oh JE, Song E, Moriyama M, et al.:
Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA.
Sci. Immunol.
2021 Dec 10; 6(66): eabj5129. Epub 20211210. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 417.
Tan HX, Juno JA, Esterbauer R, et al.:
Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles.
Sci. Immunol.
2022 Jan 28; 7(67): eabf5314. Epub 20220128. PubMed Abstract
| Publisher Full Text
- 418.
Aihara F, Wang Y, Belkina AC, et al.:
Diversity of B Cell Populations and Ig Repertoire in Human Lungs.
J. Immunol.
2023 Aug 1; 211(3): 486–496. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 419.
Pollastro S, Klarenbeek PL, Doorenspleet ME, et al.:
Non-response to rituximab therapy in rheumatoid arthritis is associated with incomplete disruption of the B cell receptor repertoire.
Ann. Rheum. Dis.
2019 Oct; 78(10): 1339–1345. Epub 20190619. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 420.
Mackensen A, Muller F, Mougiakakos D, et al.:
Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus.
Nat. Med.
2022 Oct; 28(10): 2124–2132. Epub 20220915. PubMed Abstract
| Publisher Full Text
- 421.
Mahevas M, Azzaoui I, Crickx E, et al.:
Efficacy, safety and immunological profile of combining rituximab with belimumab for adults with persistent or chronic immune thrombocytopenia: results from a prospective phase 2b trial.
Haematologica.
2021 Sep 1; 106(9): 2449–2457. Epub 20210901. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 422.
Mariette X, Barone F, Baldini C, et al.:
A randomized, phase II study of sequential belimumab and rituximab in primary Sjogren’s syndrome.
JCI Insight.
2022 Dec 8; 7(23). Epub 20221208. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 423.
Shipa M, Embleton-Thirsk A, Parvaz M, et al.:
Effectiveness of Belimumab After Rituximab in Systemic Lupus Erythematosus: A Randomized Controlled Trial.
Ann. Intern. Med.
2021 Dec; 174(12): 1647–1657. Epub 20211026. PubMed Abstract
| Publisher Full Text
- 424.
Cavalli G, Dinarello CA:
Anakinra Therapy for Non-cancer Inflammatory Diseases.
Front. Pharmacol.
2018; 9: 1157. Epub 20181106. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 425.
Biesemann N, Margerie D, Asbrand C, et al.:
Additive efficacy of a bispecific anti-TNF/IL-6 nanobody compound in translational models of rheumatoid arthritis.
Sci. Transl. Med.
2023 Feb; 15(681): eabq4419. Epub 20230201. PubMed Abstract
| Publisher Full Text
- 426.
Lagares D, Santos A, Grasberger PE, et al.:
Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis.
Sci. Transl. Med.
2017 Dec 13; 9(420). PubMed Abstract
| Publisher Full Text
| Free Full Text
- 427.
Aghajanian H, Kimura T, Rurik JG, et al.:
Targeting cardiac fibrosis with engineered T cells.
Nature.
2019 Sep; 573(7774): 430–433. Epub 20190911. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 428.
Granit V, Benatar M, Kurtoglu M, et al.:
Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study.
Lancet Neurol.
2023 Jul; 22(7): 578–590. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 429.
Huang H, Wang Z, Zhang Y, et al.:
Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer.
Cancer Cell.
2022 Jun 13; 40(6): 656–673.e7. Epub 20220505. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)