Keywords
Escherichia coli, enteroaggregative, SepA, biofilm,
Escherichia coli, enteroaggregative, SepA, biofilm,
Enteroaggregative Escherichia coli (EAEC) is associated with acute and chronic diarrhea worldwide as well as travelers’ diarrhea.1–10 Populations strongly affected by EAEC include children in developing countries and deployed military personnel.1,9,11,12 Globally, diarrhea is the second leading cause of death in children under five.13 Among deployed military personnel, EAEC is the second most common cause of travelers’ diarrhea, a condition that is the primary generator of lost person-hours and reduced operational readiness.8,9,14–16 EAEC creates thick biofilms on the intestinal mucosa.1,17–20 Biofilm formation is thought to be necessary for EAEC to cause diarrhea.1,17–20 EAEC was first characterized by a stacked-brick pattern of adherence and aggregation on HEp-2 cells.17,19,21 Currently EAEC are identified by screening for the gene for the positive regulator of virulence genes, aggR, and genes such as those for the aggregative adherence fimbriae (AAF).22–24 EAEC strains are heterogeneous, and may have a variety of virulence genes.
In addition to aggR and genes for AAF, EAEC usually encodes one or more serine protease autotransporter of Enterobacteriaceae (SPATE).25 SepA is a SPATE that is found in 21-60% of EAEC, depending on the study.5,26–30 Epidemiological data indicate that SepA may be important for disease caused by EAEC. For example, data from Mali collected during the MAL-ED study showed that sepA was the EAEC gene most strongly associated with diarrheal cases among children under five years of age.26 In a similar analysis of Gambian data, sepA was also associated with childhood diarrheal cases.3 A study in Thailand on EAEC-mediated diarrhea in children found that sepA was one of the genes detected in significantly more cases than controls.31 A study in Iran on EAEC-mediated diarrhea in children found that isolates positive by PCR for agg4A (AAF type 4), and the SPATE genes pic and sepA formed stronger biofilm in vitro than strains without those genes.32 The AAF are important for EAEC biofilm formation and adherence.21,28,33–37
Despite the implications of these epidemiological studies, little is known about the function of SepA. All SPATEs mediate their own secretion, contain a secreted serine protease domain, and are thought to have a unique protease target. Currently, the target of the serine proteinase activity of SepA for EAEC is unknown.38 In Shigella, deletion of sepA reduces fluid accumulation in the rabbit ligated ileal loop model.39 However, unlike Shigella, EAEC is not an invasive pathogen; therefore, SepA may play a different role in EAEC pathogenesis than it does for Shigella.
In our collection of EAEC isolates from the Trial Evaluating Ambulatory Therapy of Travelers’ Diarrhea (TrEAT-TD) study, which investigated travelers’ diarrhea in UK and US military personnel deployed in Kenya, Djibouti, Afghanistan, and Honduras,40 23% of the strains positive for AAF were positive for sepA. To investigate the role of SepA in EAEC pathogenesis, we deleted sepA from EAEC strain K261. We found that K261ΔsepA exhibited increased biofilm staining in vitro. Therefore, we deleted sepA from several additional strains. For some strains the sepA49-4043 deletion caused increased biofilm staining, but for others the sepA mutant strain had equivalent biofilm to the wild-type (wt) parental strain. Ultimately, complementation of the sepA mutation, either genetically or by adding SepA exogenously, did not restore wt biofilm staining. Our results suggest that the reason for the elevated biofilm in some EAEC sepA mutant strains is not directly related to the lack of SepA.
Bacterial strains used in this study are listed in Table 1. We selected sepA-positive strains from our collection of EAEC clinical isolates obtained from the Trial Evaluating Ambulatory Therapy of Travelers’ Diarrhea (TrEAT-TD) study, which investigated travelers’ diarrhea in UK and US military personnel deployed in Kenya, Djibouti, Afghanistan, and Honduras.28,40 For an agg5A-positive, sepA-positive strain, we used D5613 (C267-15), a strain isolated from a pediatric case in Mozambique.29 HS is a commensal E. coli strain.41,42 We defined EAEC as having both aggR (EAEC virulence gene regulator43–46) and the genes for production of an AAF. The constructs used for this study are listed in Table 2.
| Strain | Aggregative adherence fimbriae (AAF) | Adhesin | Dispersin† | Regulators | aaiC‡ | SPATEs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aggA AAF1 | agg4A AAF4 | agg5A AAF5 | hra1 | aap | aatA | aar | aggR | pic | sat | sepA | ||
| P433 | + | + | + | + | + | + | + | + | ||||
| K261 | + | + | + | + | + | + | + | + | ||||
| E161 | + | + | + | + | + | + | + | + | ||||
| K411 | + | + | + | + | + | + | ||||||
| E131 | + | + | + | + | + | + | + | + | ||||
| D5613 | + | + | + | + | + | + | + | + | + | |||
# Whole genome sequence contigs published in28,29 were queried with the Center for Genomic Epidemiology’s online Virulence Finder, Serotype Finder, and Plasmid Finder platforms. In this table, only virulence genes found in one or more strains are shown.
‡ aaiC, located on the chromosome, encodes a secreted type VI secretion system effector protein.72
| Name | Description | Source |
|---|---|---|
| pKD46spec | A repA101 plasmid that confers temperature sensitivity and spectinomycin resistance with L-arabinose-inducible lambda Red protein expression under control of the araBAD promoter. Generated from pKD46amp.47 | 48 |
| pAA | Name for the EAEC virulence plasmid | This study |
| pAAΔsepA | The EAEC virulence plasmid with sepA49-4043 deleted and replaced with cat, ΔsepA::cat | This study |
| pAAP433-ΔsepA | As above, the ΔsepA::cat mutation was made in the P433 strain background. | This study |
| pAA-ΔsepA::catrev | The EAEC virulence plasmid with sepA4-4091 deleted, retaining only the start and stop codons and cat inserted with the promoter in the opposite direction from the sepA promoter. | This study |
| pTrcHis2::sepA | The wt passenger domain of sepA from K261 in the inducible vector pTrcHis2c. Expression of SepA from this plasmid would result in a SepA passenger domain with six histidines added at the N terminus. | This study |
| pTrcHis2::sepA* | The passenger domain of sepA with the S267A mutation in the inducible vector pTrcHis2c. Expression of SepA* from this plasmid would result in a SepA* passenger domain with six histidines added at the N terminus. | This study |
| TOPO::sepA | pCR2.1TOPO, a high-copy number vector with wt K411 sepA and 500 bp upstream | This study |
| TOPO::sepA* | As above but with the active site of SepA inactivated by a S267A mutation (a799g & g800c) | This study |
| TOPO::sepA5251A>V | pCR2.1TOPO with the wt P433 sepA and 500 bp upstream. Note that sepAP433 has a A5251V mutation respective to K261 and K411 | This study |
| TOPOΔlacZ::sepA kan | pCR2.1TOPO with the PCR product for lambda Red-mediated replacement of lacZ with wt sepA and kan | This study |
| pACYC177 | A low copy number vector previously described | 73,74 |
| pACYC177:sepA | sepA and 500bp upstream of the start codon (from K261) from TOPO::sepA digested with BamHI and inserted in the low copy vector pACYC177 | This study |
| sepA-kan pAAK261 | The K261 pAA with kan resistance gene inserted 200 bp downstream of sepA | This study |
| pAAK411-revertant | sepAK261-kan was inserted into pAAK411-ΔsepA to create K411Δcat:: sepAK261-kan | This study |
| TOPO::t’ase-sepA-t’ase | pCR2.1TOPO with a 6,590 bp region of sepA including the putative transposases (t’ase) upstream and downstream that were conserved for K261, K411, P433 | This study |
| TOPO::t’ase-ΔsepA::cat-t’ase | pCR2.1TOPO with the corresponding 3,360bp region of ΔsepA::cat including the putative transposase upstream and downstream that were conserved for K261, K411, P433 | This study |
Bacterial cultures were grown in a shaking incubator at 37°C with appropriate antibiotics, and diluted in Dulbecco’s Modified Eagle Medium (DMEM) with high glucose and L-glutamine (Genesee Scientific 25-501) to 107 CFU/mL as estimated by optical density (OD). For those assays, 180 μL of the culture was added to a 96-well flat-bottom untreated plate (VWR 82050-760). DMEM was used as the control for each plate. After covering the plate with a lid, the plate was incubated at 37°C without shaking for the time indicated in the figure legend. After growth, the media on top of the biofilm was removed, and the biofilm was washed once with phosphate-buffered saline (PBS) (Fisher Scientific 70-011-044) before fixing with ethanol for 10 minutes. The dry biofilm plate was stained by immersing in a mixture of 3 mM crystal violet (Sigma Aldrich C0775), 5% ethanol, and water for 30 minutes then rinsed in tap water and dried. The bound crystal violet was eluted with 100 μL ethanol in each well, and the absorbance read at 590 nm. The control DMEM-only well values were subtracted from the absorbance values for all other wells.
To assess biofilm formation on glass disks, a single sterilized glass coverslip (Fisher 12-545-81P) was placed at the bottom of each well of a 24-well plate. Each well was then inoculated with 500 μL of 107 CFU/mL bacteria in DMEM, and then otherwise treated identically to the 96-well plate biofilm method. After fixing and crystal violet staining, each disk was removed and glued to a glass slide with Cytoseal XYL (Thermo Scientific 8312-4) for imaging at 100× with oil immersion using an Olympus BX60F-3.
To test addition of components to the biofilm media, we supplemented the DMEM at the start of growth to 1 mg/ml DNase (Sigma Aldrich DN-25), 1 mg/ml chymotrypsin (Sigma Aldrich C4129), or 0.8 mM - 2.5 mM sodium metaperiodate (Fluka Analytical 71859). Another set of wells had the equivalent volume of vehicle control (PBS) added. For biofilm media with 2 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma Aldrich P7626) we made a stock of 100 mM in isopropanol and added the equivalent volume of isopropanol to a set of control wells.
To make DMEM with M9 levels of magnesium (10 mM Mg2+), we grew biofilms in regular DMEM or DMEM with an extra 9.2 mM magnesium sulfate (Acros Organics 42390500).
Virulence gene deletion strains were constructed by lambda Red-mediated recombination with the pKD46spec plasmid.47,48 In most cases, the entire gene was replaced with a kanamycin (kan), gentamicin, or chloramphenicol (cat) resistance gene. Briefly, PCR primers, listed in Table 3, with approximately 50 nucleotides of homology to the wt gene were used to amplify the resistance gene. The PCR product was gel-purified following kit directions (Qiagen 28506) and electroporated into EAEC induced for expression of the lambda Red system (1 mM arabinose; Chem-Impex 01654), using a MicroPulser Electroporator set to 1.8 kilovolts with 200-ohm resistance (BioRad 1652100).49 After electroporation, the EAEC were incubated at 37°C for one hour to allow the recombination (based on homology to the primer regions) to proceed and for expression of the antibiotic resistance. Growth at 37°C also promotes loss of the pKD plasmid.48 Gene deletions were confirmed by PCR with specific screening primers listed in Table 3. We also confirmed, by plasmid sequencing, that the ΔsepA::cat mutation was the only genetic change in pAAK261-ΔsepA as compared to pAAK261 and for pAAP433-ΔsepA relative to pAAP433, and that the sequence of revertant (pAAK411-revertant) was correct.
To amplify PCR products larger than 4 kb, we used Platinum SuperFi II PCR Master mix (Invitrogen 12368010). For PCR products less than 4 kb, we used PfuUltra II Fusion HS Master mix (Agilent Technologies 600852). For confirmatory PCR from colonies, we used colony “boils” (one colony was added to 50 uL of water with a sterile toothpick and incubated for 12 min at 95°C), and the GoTaq Green Master mix kit (Fisher PRM7123).
To insert PCR products into pCR2.1TOPO (TOPO), we followed manufacturer guidelines (Invitrogen K280020). PCR products were purified using the QIAquick PCR & Gel Cleanup kit (Qiagen 28506). Plasmids were purified using the QIAprep Spin Miniprep kit (Qiagen 27106X4). To purify larger quantities of the pAA plasmid, we increased the yield of this large, low-copy vector by adding extra yeast extract to the Luria-Bertani (LB) broth (final concentration of 24 g/L yeast extract), as described by Wood et al.50 To move inserts into pACYC177, we digested the sepA inserts from the TOPO clones with BamHI (NEB R3136), then ligated the inserts into pACYC177. Primers were sourced from Integrated DNA Technologies and are listed in Table 3.
To allow the selection of colonies with sepA inserted into lacZ, we first added a kan resistance gene 200 bp downstream of sepA prior to amplification of the entire region for lambda Red-mediated replacement of the lacZ gene. We confirmed that addition of kan after sepA had no effect on biofilm staining in the wt strains (data not shown).
For the larger PCR construct, ΔlacZ::sepA-kan, after amplification using K261 pAA-kan as a template, the construct was first inserted into TOPO per manufacturer instructions. The plasmid was then digested with NotI (NEB R3189) and BamHI (NEB R3136) and gel-purified to yield 500 ng of insert DNA and added to 100uL of electrocompetent cells for lambda Red recombination. The revertant strain (K411Δcat::sepA-kan) was identified by screening for loss of chloramphenicol resistance after recombineering with the ΔlacZ::sepA-kan PCR (Table 3). We confirmed the localization of SepA in the supernatant using Western blot analysis (data not shown).
We selected a spontaneous nalidixic acid resistant (NalR) derivative of commensal E. coli HS41,42 by growth on LB agar with 15 μg/mL nalidixic acid (Sigma Aldrich N4382). We moved the K261 pAA into HSNalR by centrifuging overnight cultures of both strains, resuspending the pellets in PBS, and mixing them (HSNalR and K261) at 1:1 v:v. After 5 minutes of incubation at room temperature, 75 μL of the cell mixture was spotted onto agar without any antibiotics and incubated for three hours. The mix was then inoculated onto MacConkey agar with 15 μg/mL nalidixic acid to select for HSNalR and 15 μg/mL tetracycline (Sigma Aldrich 37919) to select for pAAK261 and grown overnight.
To get pAAP433ΔsepA, pAAK261ΔsepA, and pAAP433 into HSNalR, the plasmids were purified as described above using extra yeast extract in the LB broth medium, then 1 μg of the plasmid was transformed into electrocompetent HSNalR cells. The transformed HSNalR was grown at 37°C overnight before plating on LB agar with 30 μg/mL chloramphenicol (Calbiochem 220551) to select for uptake of the pAAs with ΔsepA or 100 μg/mL ampicillin (Corning 61-238-RH) agar for pAAP433.
In all cases, single colonies were screened by PCR to confirm the presence of aggR (pAA marker) and the absence of aaiC (EAEC chromosomal marker) to confirm uptake of the pAA plasmid.
A mutation in the active site of the SepA passenger domain (sepA*) (Table 2) was generated by splicing by overlap extension (SOE) PCR using primers SepAS211F and SepAS211R (Table 3) to create the following changes: a799g & g800c in sepA which result in replacement of the serine at position 267 with an alanine. Next, the coding region for the passenger domains of K411 sepA and sepA* were amplified by PCR from TOPO::sepA* and TOPO::sepA to add BamHI and SalI cut sites, and ligated into the inducible vector pTrcHis2c (Invitrogen V36520) (Table 2). The expression of SepA (or SepAS267A*) from these plasmids would result in SepA with a hexahistidine (His6) tag added at the N terminus. We chose to use the His tag for purification purposes because both Scott-Tucker et al. and Charbonneau et al. showed that the serine protease activity of a SPATE passenger domain is retained even with an N-terminal hexahistidine-tag.51,52 After optimizing SepA expression from TOP10 pTrcHis2c with 1 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) (Sigma Aldrich I6758), the bacterial cells were lysed with BugBuster Master mix (Novagen 71456). The protein was then purified with a HisTrap HP nickel column according to the manufacturer’s protocol (GE Healthcare 95056-204). The purity of fractions collected from the nickel column was evaluated on a 4-12% Bis-Tris gel (Invitrogen NP0324). The fractions with the 110 kDa His-SepA protein band were concentrated with an Amicon Ultra-15 centrifugal concentrator 50,000 MW (Fisher Scientific UFC905008). The same type of centrifugal concentrator was used for buffer exchange with PBS to remove the imidazole.
For harvesting K411 wt or K411ΔsepA supernatant, cultures were grown in DMEM high glucose, with L-glutamine (Genesee Scientific 25-501) at 37°C shaking for five hours prior to centrifugation. The supernatants were filtered with a 0.22 μm filter (Genesee Scientific 25-227) before concentrating 2,000-fold with an Amicon 50 kDa cutoff spin column (Fisher UFC905008). The concentrated supernatant was then added to the biofilm media at the start of incubation. For TOPO::sepA or TOPO::sepA* supernatants, the plasmid was grown in TOP10 cells (Invitrogen C404010) and harvested identically to the EAEC strains.
A peptide (K261 SepA891-913) from the exposed regions of the passenger domain of SepA was selected to generate a rabbit anti-SepA polyclonal antibody. Pacific Immunology (Ramona, CA) made rabbit polyclonal antibodies by immunizing two rabbits with the peptide at day 0 with complete Freund’s adjuvant and then again on days 21, 42, and 70 with incomplete Freund’s adjuvant. On day 77, 25 ml of serum was collected and affinity purified. This animal protocol was approved by IACUC-designated member review on August 12, 2019 from Pacific Immunology (SOP-1).
Bacterial strains (listed in the figure legends) were grown at 37°C for four hours in DMEM with shaking for liquid culture conditions or without shaking for static biofilm cultures. The supernatants from those cultures were collected after centrifugation and precipitated with an equal volume of ethanol overnight at -20°C. Samples were denatured with NuPAGE® sample buffer (Invitrogen NP0008) and electrophoresed on a 4-12% Bis-Tris gel (Invitrogen NP0324). The proteins were transferred using an iBlot nitrocellulose transfer stack (Invitrogen IB301002) to nitrocellulose. The nitrocellulose blot was incubated for two hours at room temperature in PBS with 0.1% Tween (Fisher Scientific 424592500) with 5% nonfat dry skim milk (Lab Scientific M0841). After the blocking step, the nitrocellulose was incubated overnight at 4°C with the anti-SepA antibody, at a dilution of 1:4,000 or 1:7,000 (listed in the figure legend), in PBS-Tween-milk, then washed three times with PBS-Tween. Next, the blot was incubated with a 1:10,000 dilution of the secondary antibody (goat anti-rabbit HRP conjugate; Bio-Rad 1706515) in PBS-Tween at room temperature for two hours. The nitrocellulose blot was washed once with PBS-Tween and twice with PBS. The nitrocellulose blot was incubated with ECL Western Blotting Detection reagent according to the manufacturer’s instructions (GE Healthcare 45-002-401) and imaged with a GE Amersham 680 Blot Imager. Blot images were cropped but otherwise not manipulated.
To purify pAA DNA we used the Plasmid Midi Kit (Qiagen 12143) following the manufacturer’s published protocol.53 Sequencing of the DNA for pAAP433, pAAK261-ΔsepA, pAAP433-ΔsepA, and pAAK411-revertant was done by Plasmidsaurus (Eugene, OR). Additional DNA sequences were taken from published work.28,29 A draft assembly was done with the Minimap2 plug-in54 with Geneious Prime 2023.0.455 then further refined by re-aligning all reads in Geneious Prime. Remaining ambiguous bases were resolved manually by a comparison of the sequence quality among the sequencing reads provided.
GraphPad Prism 10.0.356 was used to test significance of three or more biological replicates by unpaired t test for two groups or an ordinary one-way analysis of variance (ANOVA) with a correction for multiple comparisons. *P≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.
In our EAEC isolate collection from the TrEAT-TD study,40 all of the typical EAEC strains positive by PCR for the sepA gene were also positive for either the aggA or agg4A AAF major subunit gene. We selected six of the recently isolated clinical strains that were positive for sepA for this study (Table 1). The virulence gene profiles of these strains are found in Table 1. We deleted sepA in these strains by replacing sepA49-4043 with a chloramphenicol resistance gene (cat). We found that two EAEC strains exhibited increased biofilm staining after the deletion of sepA (Figure 1). Based on that finding, we initially hypothesized that the proteolytic activity of SepA modulated biofilm formation in a subset of EAEC by either cleaving a protein on the surface of the EAEC or protein(s) within the biofilm. Although four of the strains tested were positive for agg4A, we also tested an aggA+ strain, E131, and an agg5A+ clinical strain, D561329 (Table 1). Neither the aggA+ nor the agg5A+ ΔsepA strains showed a significant difference in biofilm staining (Figure 1). These results suggested that SepA may only affect the biofilm of some agg4A-positive strains. We confirmed that no growth defects were present in a selection of four of the strains (Figure 2).

Symbols in pink represent the values for the wt parental strain and in blue represent the corresponding ΔsepA strain. Each symbol shows the mean of four biofilm technical replicates grown for 23 hours. Error bars indicate standard error of the mean (SEM). Significance was determined using unpaired t tests for independent experiments done for each wt-mutant pair. ****P≤0.0001.

Each strain was grown in DMEM high glucose with L-glutamine with shaking in 250mL flasks with the appropriate antibiotic. The wt strain values are shown as pink squares and the corresponding ΔsepA strain with blue circles. The CFU and optical density, OD600, were measured every hour. Bars indicate standard deviation (SD) for n=3 or range for n=2. The number of biological replicates is written below each graph.
In Figure 1, four isolates had a non-significant increase in biofilm staining for the ΔsepA mutant strain as compared to the wt parental strain. Because the overall biofilm levels were relatively high for all of the strains, we reasoned that we might be able to detect a significant difference between the wt and the ΔsepA derivative strains if we reduced the amount of time that the biofilms were allowed to develop. Therefore, we assessed when we could detect the elevated biofilm staining phenotype by testing shorter growth times for biofilm growth for K261 and K261ΔsepA (Figure 3). We found that the increased biofilm staining by K261ΔsepA relative to K261 was apparent after five hours of biofilm growth and remained differentiated up to the typical 23-hour time point we used for longer biofilm assays (Figure 3). We also observed a linear relationship between increased growth time and increased biofilm staining for each strain as predicted by a linear regression model, although the lines were not perfectly parallel (Figure 3). The best separation in biofilm staining for K261 compared to K261ΔsepA was found for biofilms grown for less than nine hours (Figure 3). Based on these results, we decreased the growth time of the biofilm to five to eight hours and re-tested the EAEC strain pairs for differences in biofilm staining. With the shorter biofilm growth time, we observed a statistically significant difference in staining for E131 and E161 as compared to the corresponding ΔsepA-derivative strains when we controlled for variability by normalization (Figure 4). E131 has the aggA AAF gene (Table 1), so these results demonstrate that the ΔsepA biofilm phenotype is not limited to strains with the agg4A gene. Strains P433ΔsepA (agg4A) and D5613ΔsepA (agg5A) did not show a statistically significant change in staining compared to the wt parental strain under these conditions (Figure 4).
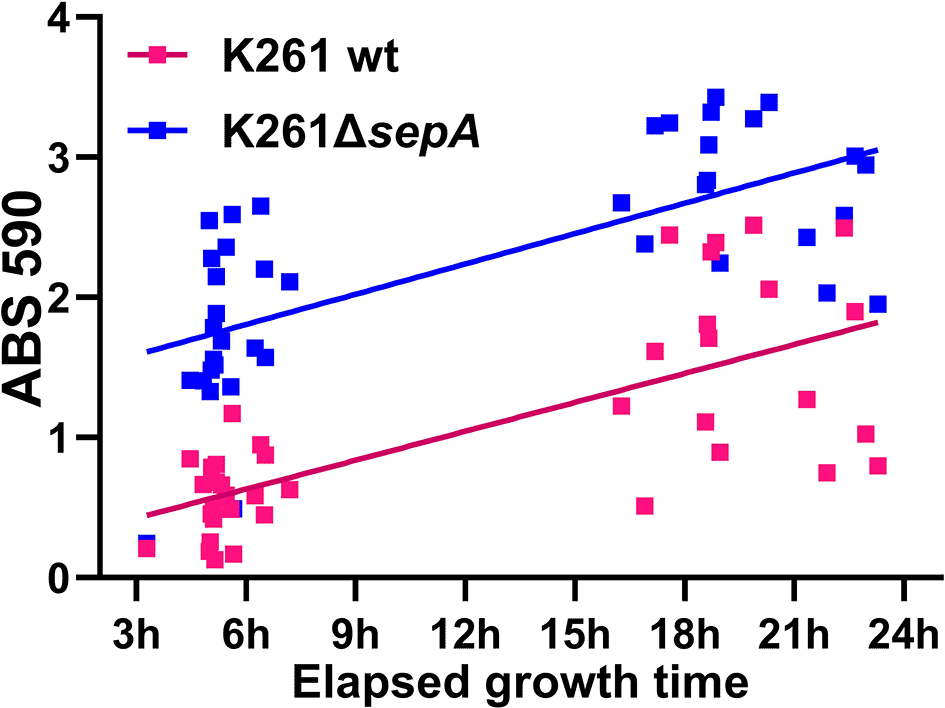
Biofilm results from 96-well plate assays. Pink symbols represent K261 and blue symbols the K261ΔsepA strain. Each symbol represents a biological replicate. The lines are a simple linear regression determined in GraphPad Prism.

The biofilm absorbance for the mutant strains was normalized to that of the wt strain. Pink symbols represent the wt parental strain and blue symbols indicate the corresponding ΔsepA strain. Each symbol represents the mean of four technical replicates. Significance was determined using an unpaired t test for independent experiments performed for each mutant-wt pair. *P≤0.05; **P≤0.01.
Since the original sepA deletion strategy left 96 nucleotides of the sepA coding region, we next made a deletion that left only the start and stop codons, and reversed the orientation of cat gene (catrev) relative to sepA. K411ΔsepA::catrev exhibited increased biofilm staining compared to the wt parental strain (Figure 5). This result indicates that the increased biofilm staining after deletion of sepA was not an artifact of the original mutation strategy.
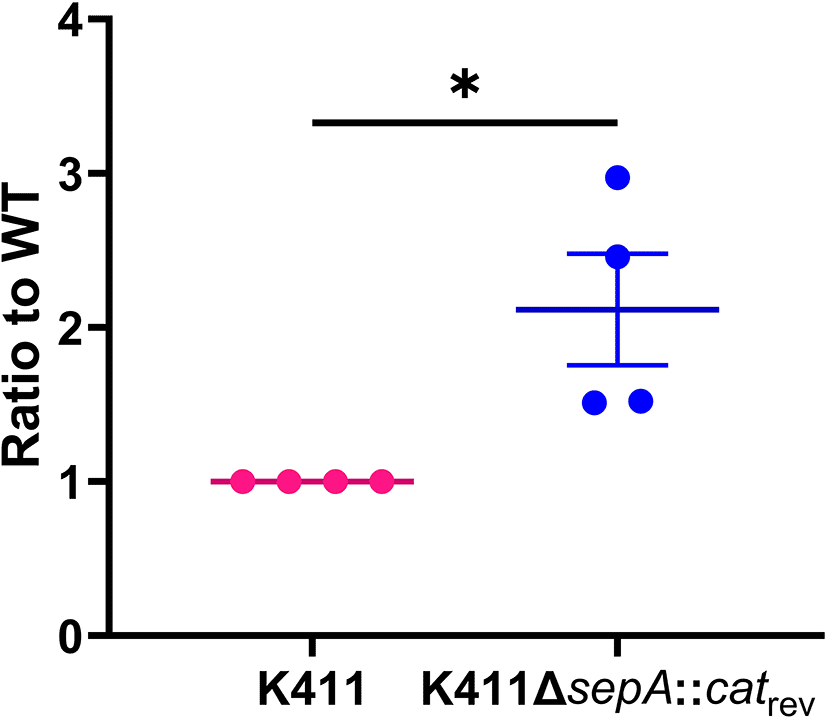
Biofilm results from 23 hour 96-well plate assays. Pink symbols indicate the wt parental strain, and blue symbols are for the corresponding ΔsepA::catrev strain. Each symbol represents the mean biofilm staining of four technical replicates. Error bars indicate SEM. P-value from unpaired t test *P≤0.05.
We hypothesized that there might be an expression level difference or an amino acid difference within SepA among different EAEC strains that prevented a SepA-mediated reduction in biofilm staining in the wt strains. We found no nucleotide differences in the 185 bp upstream of sepA, a region that includes the binding region for the EAEC virulence regulator AggR,57,58 for any of the strains in this study. This finding demonstrates that the sepA promoter is the same for all of the strains. Next, we compared the predicted amino acid sequences of SepA among the EAEC used in this study. There were several amino acid polymorphisms with respect to the predicted protein consensus derived from all strains combined: (K411: A51V), (K261: S905N), (E131: I183V, G194E, D1049N), (D5613: D1049N, H671N), (P433 & E161: A1277V). However, we observed no common amino acid change in SepA among the strains that showed a difference in biofilm staining when sepA was deleted compared to those that did not (P433 and D5613). The most interesting change identified was A1277V in E161 and P433. The A1277V change is in the predicted autotransporter beta domain, which is necessary for secretion of the proteolytic passenger domain into the supernatant. In other SPATEs, mutations in this domain can inhibit secretion of the SPATE.51,59–62 Accordingly, we examined SepA secretion into the supernatant from a subset of EAEC in this study, shown below.
To investigate whether the increased biofilm staining observed when sepA is deleted from some EAEC but not others is related to SepA secretion levels, we performed Western blot analysis on EAEC culture supernatants. We were particularly interested in whether the amino acid substitution found only in E161 and P433 (A1277V) had an impact on secretion. We observed the same apparent SepA band in all the wt strains examined, and all the strains tested appeared to have approximately equal levels of SepA (Figure 6). This finding was true for EAEC with (K411, K261, and E161) or without (P433) a difference in biofilm staining when sepA was deleted. We also examined the supernatants from statically-grown biofilms of K261 and P433 for SepA. Similar SepA bands were observed from the supernatant harvested from the biofilms of K161 and P433 (Figure 6). There was no visible SepA band identified from the bacteria contained in the cell pellet from the biofilm-grown bacteria; therefore, we inferred that SepA was efficiently exported into the supernatant for both K261 and P433 (Figure 6).
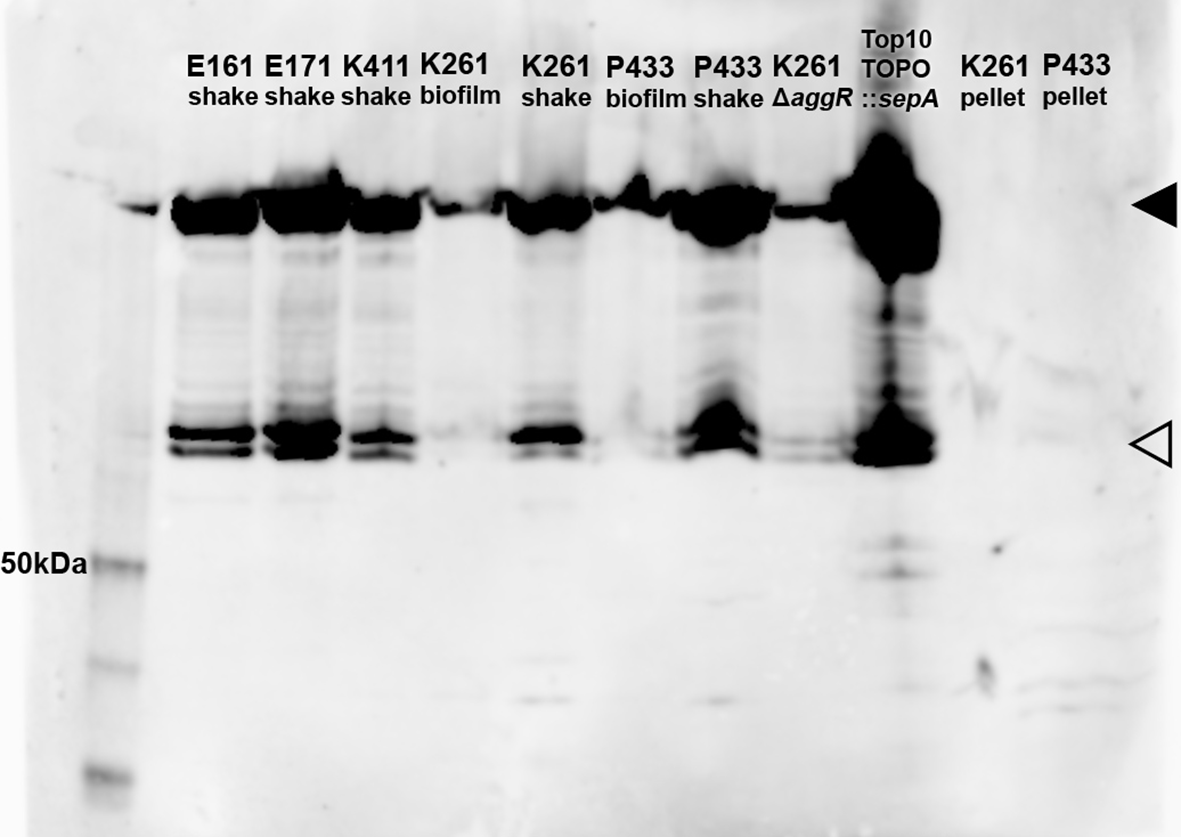
Supernatants from shaking (shake) cultures, a static biofilm culture (biofilm), or cell pellets are shown. The black triangle (◂) denotes the band corresponding to the SepA passenger domain, at approximately 110 kDa39 while the open triangle (⊲) represents an unknown breakdown product. The first lane is the MagicMark XP standard (Invitrogen LC5602) with the 50 kDa band indicated. E171 is a wt sepA+ EAEC strain that is genetically identical to E16128 (not used elsewhere in this study).28 1:4,000 anti-SepA antibody.
We also tested the impact of deletion of the EAEC virulence gene regulator, aggR (K261ΔaggR) on SepA secretion. AggR is known to positively regulate sepA expression via an AggR-binding site.57,58 We found that some SepA was still secreted when aggR was deleted (Figure 6). We also found that SepA was efficiently secreted by a laboratory strain carrying a high-copy plasmid containing the K261 sepA gene (which was inserted in the opposite orientation as the lac promoter), without AggR (TOPO::sepA). This plasmid was later used as a part of our complementation work.
To visualize the biofilm after sepA was deleted, EAEC were grown on glass disks under the same conditions used for the 96-well biofilm assay (Figure 7). We examined K261 (showed elevated biofilm staining as measured by OD when sepA was deleted) and P433 (no change in biofilm staining OD when sepA was deleted). We observed that the ΔsepA strain biofilm patterns were different than found in the wt strains. Both P433 and K261 appeared to be packed closer together than the respective ΔsepA strains (Figure 7). However, this difference in biofilm appearance does not correlate to the quantitative amount of crystal violet staining, for reasons that are not clear.

Biofilms were grown for five hours on glass disks and stained with crystal violet. Representative images from two independent experiments are shown at 100× magnification.
We next sought to determine whether the differences in biofilm crystal violet staining for K261 and K261ΔsepA were due to differences in the bacterial numbers in the biofilm. We checked both the biofilm and biofilm supernatant for K261 and, for comparison, P433 as well as the corresponding ΔsepA strains. No differences in CFU were identified (Figure 8). As the CFU from biofilms for K261, P433, and the ΔsepA derivative strains did not deviate, the staining differences were not due to reductions in bacterial numbers.
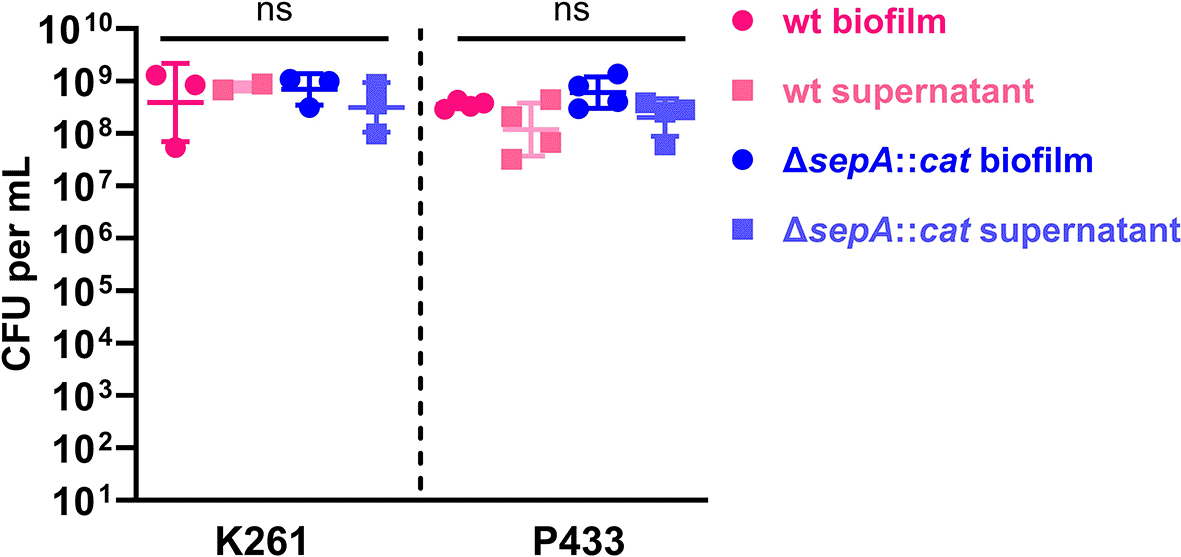
Biofilms were grown as usual in 96-well plates for five hours. Subsequently, 100 μL of supernatant was collected for CFU enumeration (supernatant). After washing the biofilm once with PBS, the biofilm was removed by scraping and resuspended in 100 μL of PBS and vortexed (biofilm). To confirm separation of bacterial aggregates after vortexing, we checked the mixture by microscopy (data not shown). Crystal violet staining after biofilm removal confirmed that no biofilm remained in the wells after scraping. Each symbol represents the mean of two technical replicates. Error bars indicate SEM. ns, no significant differences were found using 1-way analysis of variance (ANOVA) with Tukey’s test.
To identify the genes that might be responsible for the increased biofilm staining for K261ΔsepA, but not for P433ΔsepA, we transferred the pAAs from those strains into the commensal E. coli HSNalR which does not have the capacity to make biofilm. We successfully transferred pAAK261 into HSNalR. We found that the K261 pAA did not confer biofilm formation on HSNalR (Figure 9). That result is similar to those of Alves et al. who reported that the wt pAA plasmid from EAEC strain 042 does not confer the capacity to form biofilm to an E. coli K12 strain.63 In contrast, HSNalRpAAK261ΔsepA demonstrated significantly increased biofilm staining (Figure 9). This latter result indicated that the target of SepA is likely on the K261 pAA. We also put pAAp433 and pAAP433ΔsepA (does not confer elevated biofilm to P433) into HSNalR. Biofilm staining was not increased in HSNalR with either pAAP433 or pAAP433ΔsepA (Figure 9). We also found that sepA alone or ΔsepA::cat alone did not confer the capacity to form a biofilm on HSNalR (data not shown). Therefore, we concluded that the ΔsepA::cat mutation alone is not sufficient to cause the increased biofilm staining of HSNalR that had pAAK261ΔsepA.
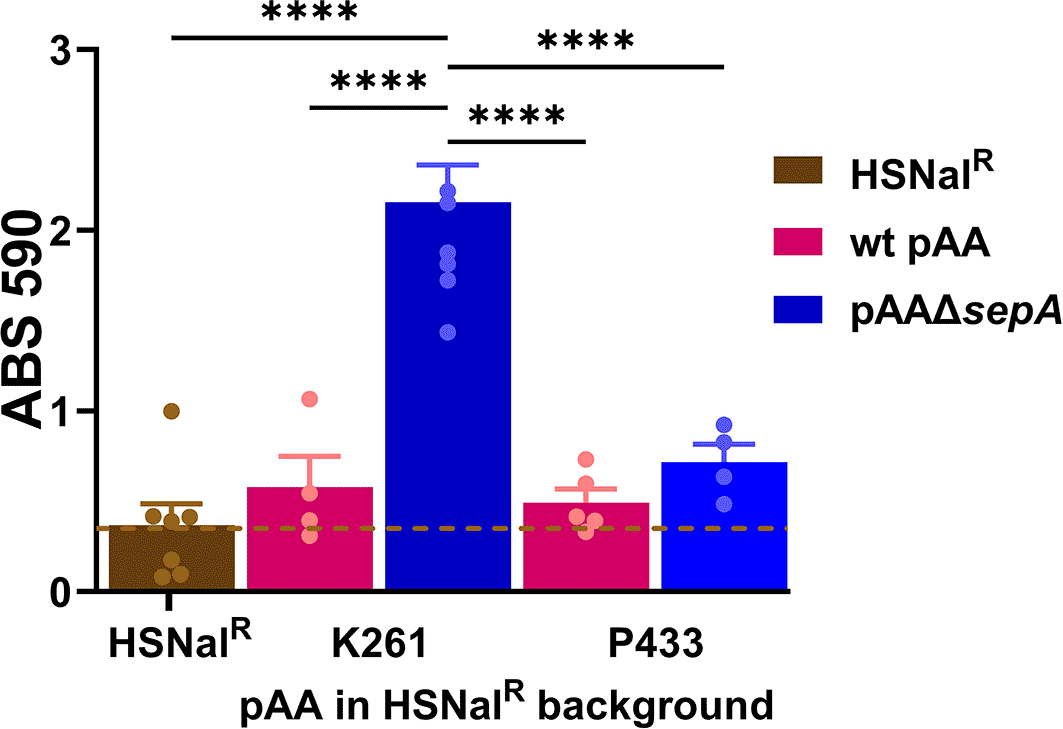
Biofilms were grown for 4.5 hours. Pink bars represent HSNalR with a wt pAA. Blue bars indicate HSNalR with a pAAΔsepA. Each symbol represents the mean of four technical replicates. Error bars indicate SEM. Significance was tested using 1-way ANOVA with Tukey’s test. ****P≤0.0001.
Because it is likely that the target of SepA is encoded on the K261 pAA, and we predicted that deleting the gene for a target of SepA would result in a significant change in biofilm staining, we searched for genetic differences in the pAAP433 as compared to pAAK261. From that search, we found that the pAAP433 lacks most of the tra (plasmid transfer) genes. Therefore, we deleted the tra region from pAAK261 (pAAΔpsiB-orf8) and assessed biofilm formation (Figure 10). Because we found no significant difference in biofilm staining between K261 and K261pAAΔpsiB-orf8, we concluded that the target for SepA was not encoded within the deleted region. Other gene deletions made in pAAK411, ΔumuC-yccA, ΔfinO, ΔparB similarly had no impact on biofilm staining (data not shown). We also searched for genes (excluding transposes) on the pAA for predicted amino acid differences found only in P433 and D5613. Excluding the previously deleted genes and mobile genetic elements, we identified lpxM (Lipid A biosynthesis myristoyltransferase)64 for which there were predicted amino acid differences between K261 and P433. However, because we found no impact of lpxM deletion on biofilm staining of K261 (Figure 10B), we concluded that LpxM is not the target of SepA.

Biofilms were grown for 5-8 hours. Each symbol represents the mean of four technical replicates. Significance tested by 1-way ANOVA with Šídák correction. *P≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.
Next, because deletion of the gene for dispersin (aap) increases biofilm staining for EAEC strain 042,65,66 we tested a strain with a deletion of both aatA (encodes the dispersin transporter) and sepA. The positively-charged Aap protein is thought to help the AAF extend from the surface of the bacteria.65,66 In Figure 7 we noted that ΔsepA bacteria in the biofilm were less compactly spaced than the wt, so we predicted that preventing export of dispersin would have the opposite effect. However, we found that biofilm formation for K261ΔsepAΔaatA was increased, a result that suggests that dispersin is not a target for SepA (Figure 10C). Next, we deleted agg4A, the gene that encodes the AAF major pilin subunit in the wt and ΔsepA background of K261 (Figure 10C). We measured a significant reduction in biofilm staining for both strains, as would be predicted for loss of the AAF.35 Because biofilm formation by the K261Δagg4AΔsepA double deletion strain was indistinguishable from K261Δagg4A, we cannot determine whether SepA has an impact on the AAF by this method. However, because the predicted amino acid sequence of Agg4A from K261 and P433 are identical, we hypothesized that Agg4A is not the target of SepA for K261. We next moved on to other strategies to identify how SepA affects biofilm staining in some of our strains.
We conducted several additional experiments to investigate potential mechanisms by which biofilm staining was increased after deletion of sepA. Because Arenas et al. showed that deletion of a SPATE in Neisseria led to increased biofilm via a surface charge change and eDNA (extracellular DNA) production,67 we asked if a surface charge difference could be observed in the EAEC strains that showed differential biofilm formation. In contrast to Arenas et al. we found that differences in cytochrome c binding (a proxy for surface charge) did not correlate with biofilm staining levels (Figure 11). For example, K261ΔsepA had decreased cytochrome c binding compared to K261, but K411 and K411ΔsepA had no significant difference in cytochrome c binding (Figure 11). For our next set of experiments, we attempted to manipulate the biofilm by addition of various compounds to the medium, Figure 12. (A) To pursue the hypothesis that there might be increased eDNA present in the biofilm of strains with elevated biofilm staining, we tested the impact of DNase I on biofilm formation. We could detect no impact on biofilm staining when biofilms were grown with 1 mg/ml DNase I (Figure 12A). (B) Next, we asked whether treatment of the biofilm with an exogenous serine protease, chymotrypsin, would alter biofilm formation. We used a serine protease because SepA is a serine protease and we reasoned that it was possible that a broad-spectrum serine protease might be able to act similarly to SepA. However, no impact on the biofilm staining was observed in the strains that were treated with chymotrypsin (Figure 12B). (C) We tested the effects of sodium metaperiodate, a carbohydrate cleaving agent. Sodium metaperiodate had no impact on biofilm staining (Figure 12C). (D) Because SepA is a serine protease, we added phenylmethylsulfonyl fluoride (PMSF), a protease inhibitor, to the biofilm. We did not observe an impact on biofilm staining when 2mM PMSF was added to the biofilm compared to the vehicle control (isopropanol) (Figure 12D). This may be because PMSF is neutralized by other biofilm components over four hours, because the protease activity of SepA is not responsible for the ΔsepA phenotype, or because PMSF does not inhibit SepA under the conditions tested. (E) Because we observed increased biofilm staining (data not shown) when EAEC were grown in M9 medium (has higher levels of magnesium than DMEM) and because magnesium affects the expression of genes on the large plasmid in Shigella,64 we supplemented DMEM to magnesium levels (10 mM Mg2+) equivalent to that in M9. However, as shown in Figure 12E, Mg2+ supplementation did not change biofilm staining significantly. Nor did magnesium lower the increased biofilm staining of K261ΔsepA (Figure 12E). In summary, we did not observe a difference in biofilm response between the wt and ΔsepA strains for any of the tested treatments.
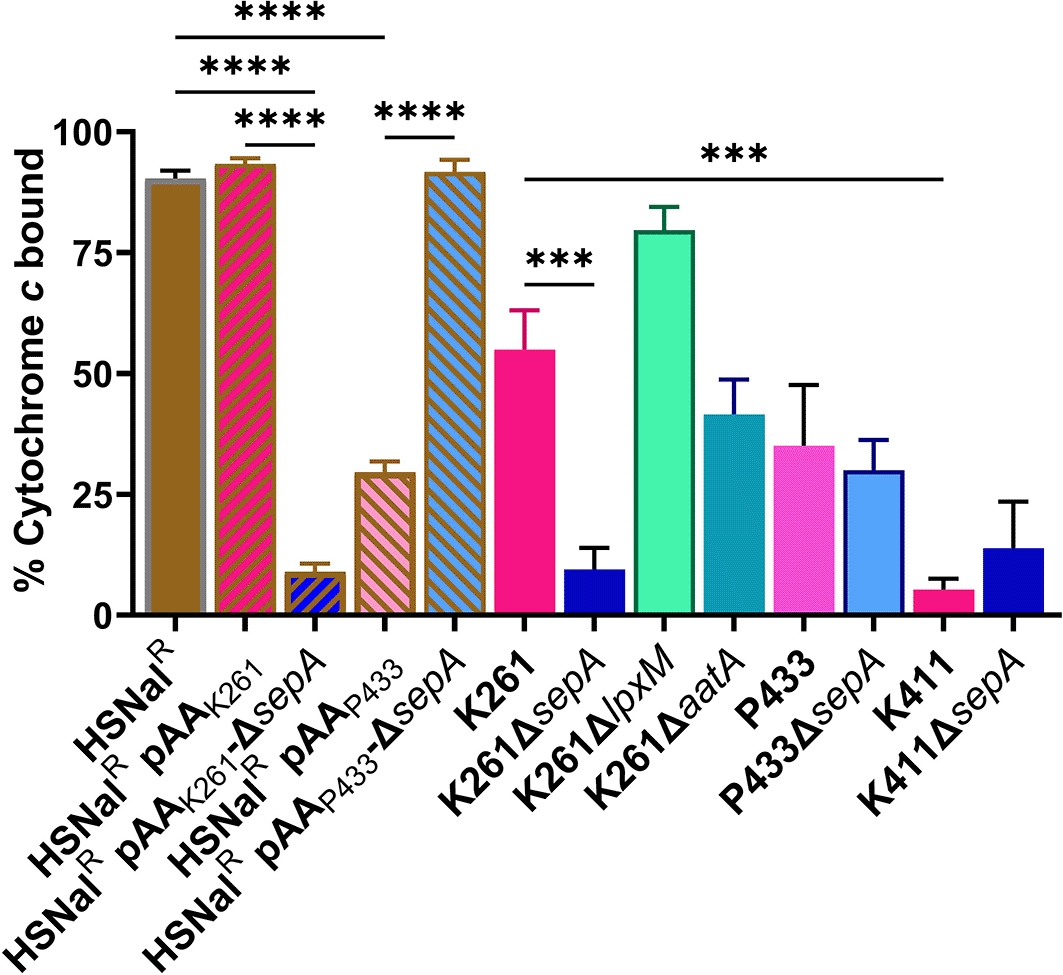
The percentage of cytochrome c bound for each strain was calculated as previously described.75 Each bar represents the mean of three biological replicates. Error bars indicate SEM. Significance tested by 1-way ANOVA with Šídák correction. ***P≤0.001; ****P≤0.0001.

Biofilm results from a five hour 96-well plate assay grown in DMEM media supplemented with (A) 1 mg/ml DNase, (B) 1 mg/ml chymotrypsin, (C) 0.8 mM – 2.5 mM sodium metaperiodate, (D) 2 mM PMSF, or (E) 10 mM Mg2+ as compared to DMEM (0.8 mM Mg2+). Pink symbols indicate K261, blue symbols K261ΔsepA, purple symbols P433, and green symbols P433ΔsepA. Square symbols represent strains with the vehicle control added to the biofilm, and circles the biofilms with supplemented media. Each symbol represents the mean of four technical replicates. Error bars indicate SEM. Significance was tested by 1-way ANOVA with Šídák correction. *P≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.
We hypothesized that a SepA-secreting strain should reduce the biofilm staining of K261ΔsepA to K261 levels. We therefore inoculated a biofilm plate with a mixture of K261ΔsepA and P433 (SepA secreter) or P433ΔsepA as a control. We selected P433 as the SepA-secreting strain because P433 and P433ΔsepA showed identical staining. Co-culture of P433 with K261ΔsepA did not significantly lower biofilm staining as compared to the P433ΔsepA (SepA non-secretor) (Figure 13). There are several possible explanations for this observation. One, SepA only acts quite close to the surface of the bacteria, or similarly, is trapped within the biofilm. Second, that SepA is rapidly inactivated in the biofilm media, or, lastly, that SepA is not responsible for the increased crystal violet staining observed in K261ΔsepA.
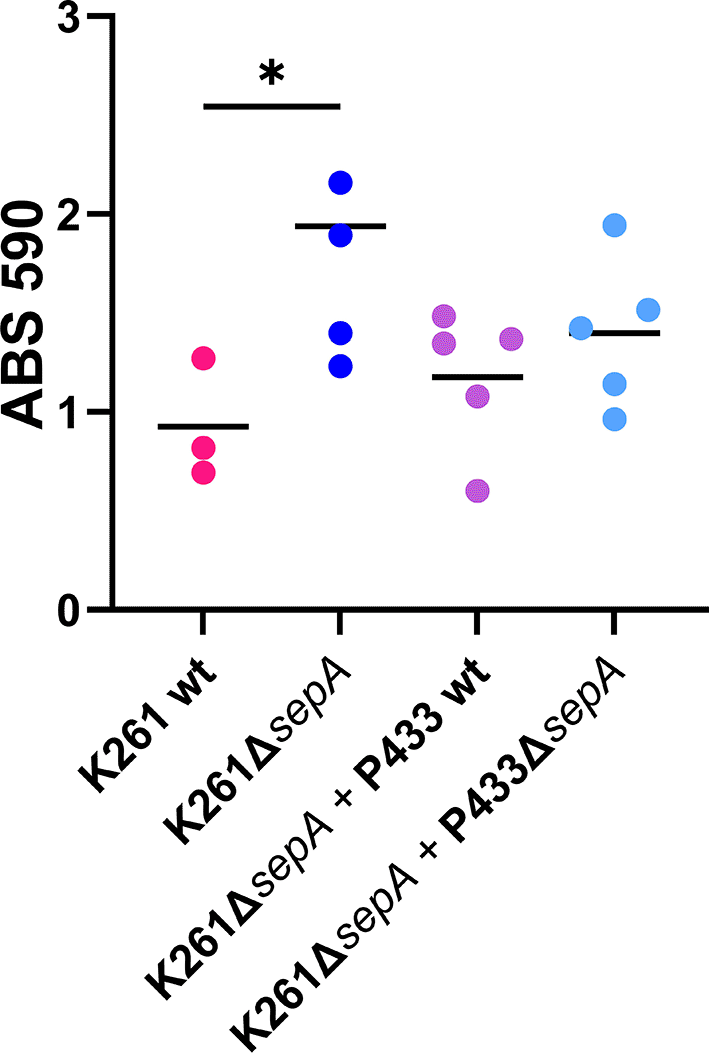
Equal volumes of the indicated strains were grown for five hours in a biofilm. Each symbol represents the mean of four technical replicates. Significance was tested using 1-way ANOVA with Šídák correction. *P≤0.05.
We next added SepA directly to the biofilm. The SepA passenger domain is abundantly secreted into the supernatant for both EAEC and lab E. coli strains (Figure 6 and Ref. 39). We chose K411 as the donor for the supernatant because it contains only one SPATE, SepA. We observed a significant reduction in staining for the K261ΔsepA biofilm grown with K411 supernatant (Figure 14A). These results suggested that exogenously added SepA could reduce biofilm staining. Although the K411ΔsepA supernatant also slightly reduced K261ΔsepA biofilm staining (Figure 14A), the reduction was not statistically significant. It may be that there were other factors present in the concentrated supernatant from K411ΔsepA, including waste products, which may impact biofilm growth.

(A) Concentrated supernatants from either K411 or K411ΔsepA were added to the growing biofilm of K261 or K261ΔsepA at the start of incubation (5-hour biofilm). (B) Concentrated supernatant from TOP10 cells with either TOPO::sepA or TOPO::sepA* was added to the growing biofilm. Absorbance was normalized to that of the untreated ΔsepA strain. Each symbol represents the mean of four technical replicates. Error bars indicate SEM. Significance tested by 1-way ANOVA with Šídák correction. *P≤0.05; **P≤0.01.
To eliminate the possible influence of other EAEC elements within the supernatants, we concentrated the supernatant fraction from laboratory E. coli TOP10 cells that carried TOPO::sepA (wt SepA protein) or TOPO::sepA* (catalytically inactive SepA). The concentrated supernatant fractions were then added to the growing biofilms as before. The presence of the supernatant from the TOPO::sepA construct did not lead to a statistically significant reduction in biofilm staining (Figure 14B), despite the abundant presence of SepA in supernatants from that construct as shown by Western blot (Figure 6).
As the concentrated supernatant from the TOP10 cells with either TOPO::sepA or TOPO::sepA* did not reduce biofilm formation, but the concentrated supernatant from K411 did, we next asked whether purified SepA protein could function to reduce biofilm staining for K261ΔsepA. We expressed the His-tagged SepA passenger domain (His-SepA) as previously described for other SPATEs.25,68,69 The catalytically inactive form of the SepA passenger domain (SepA*) was used as a control. The vehicle control (PBS), His-SepA, or His-SepA* was added to K261ΔsepA at the start of inoculation for the biofilm and again one hour before fixing the biofilm. As shown in Figure 15, no impact on biofilm staining was observed. This result, taken with mixed biofilm data, further suggested that the SepA protein may not be responsible for the difference between wt and ΔsepA biofilm staining. We were unable to measure the enzymatic activity of the His-SepA in this study due to the lack of a known substrate for the EAEC SepA. However, the utilized purification process has successfully been used in other studies to yield functional SPATEs.51,52,68 Following the outcomes of these studies, we next utilized a genetic approach to test SepA.
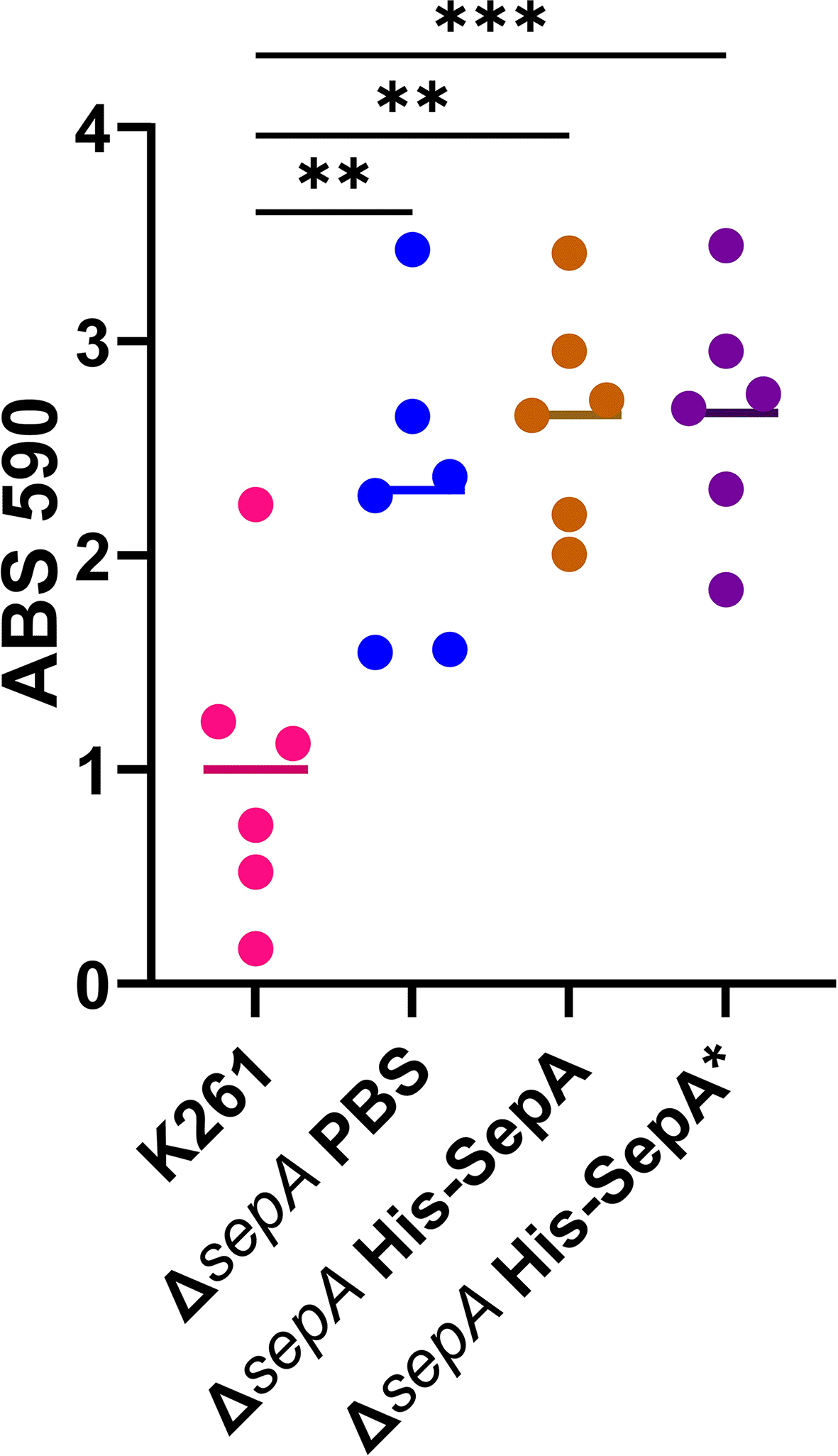
Biofilm results from a five hour 96-well plate biofilm assay are shown. Protein or PBS vehicle control was added at the time of biofilm inoculation and again one hour before biofilm fixation. Each symbol represents the mean of four technical replicates. Significance tested by a 1-way ANOVA with Tukey’s test. **P≤0.01; ***P≤0.001.
To complement the ΔsepA mutation genetically, we transformed K261ΔsepA and K411ΔsepA with pACYC177::sepA for complementation (Figure 16). We confirmed SepA expression in the supernatant from pACYC177::sepA (Figure 17) and that there was no growth defect for ΔsepA strains with pACYC177::sepA (data not shown). However, this construct failed to reduce biofilm staining of K261ΔsepA and K411ΔsepA as compared to the parental strains (Figure 16).
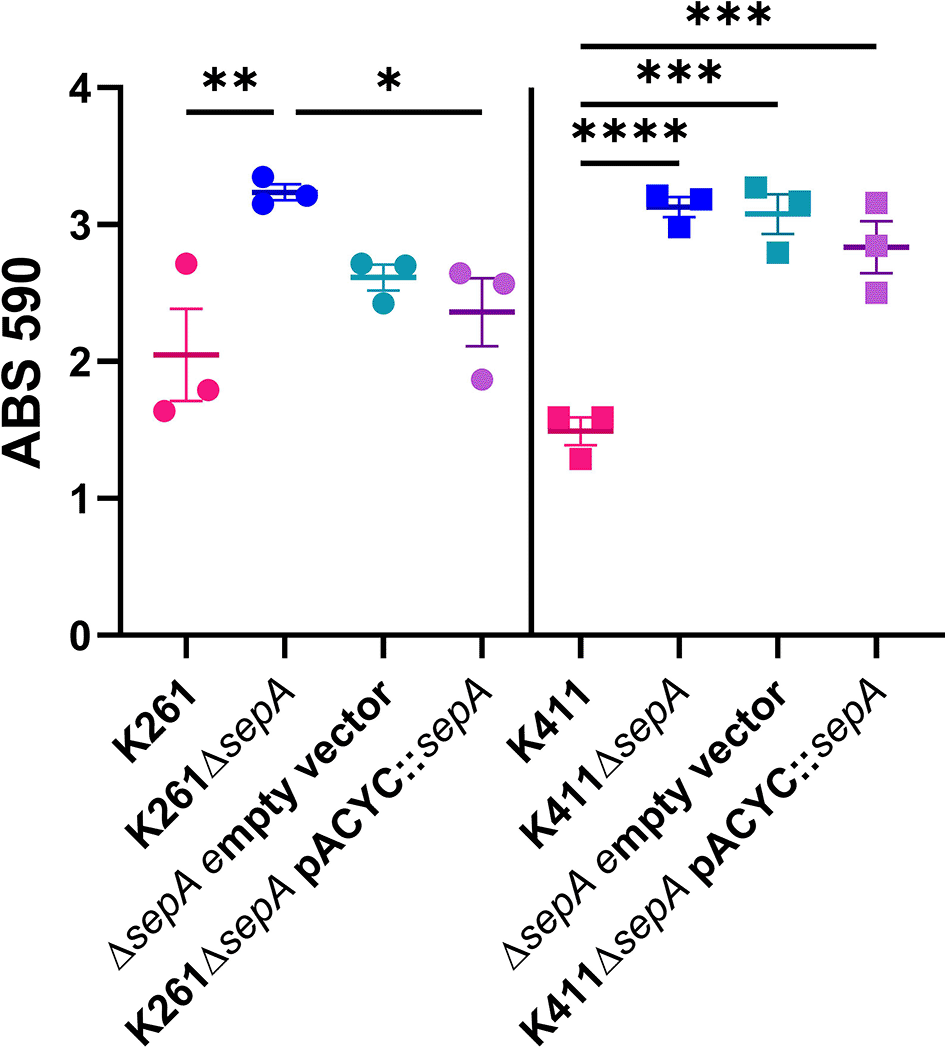
Biofilms were grown for 23 hours in a 96-well plate. Each symbol represents the mean of four technical replicates. Error bars indicate SEM. Significance was tested by 1-way ANOVA with Šídák correction. *P≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.

Supernatants from four hour shaking cultures of the indicated strains. 1:7,000 dilution of the SepA antibody. Black triangle (◂) points to the SepA passenger domain band. Open triangle (⊲) indicates an unknown breakdown product band also seen in Figure 6.
Since the plasmid-based methods failed to complement the altered biofilm-staining phenotype in the ΔsepA strains, we next introduced sepA into the chromosome of K411ΔsepA, in the lacZ locus. However, we did not observe a reduction in biofilm staining of K411ΔsepAΔlacZ::sepA (Figure 18). In our last attempt at complementation, we created a “revertant” strain in which sepA was inserted back onto the pAA in place of the deleted sepA. The revertant strain had equivalent biofilm staining as found for the ΔsepA strain (Figure 18), a finding that demonstrated that the reason for the elevated biofilm staining in this strain is not due to the sepA deletion. Collectively, these data suggest that the lack of SepA did not cause the increase in the biofilm formation in the ΔsepA strains.
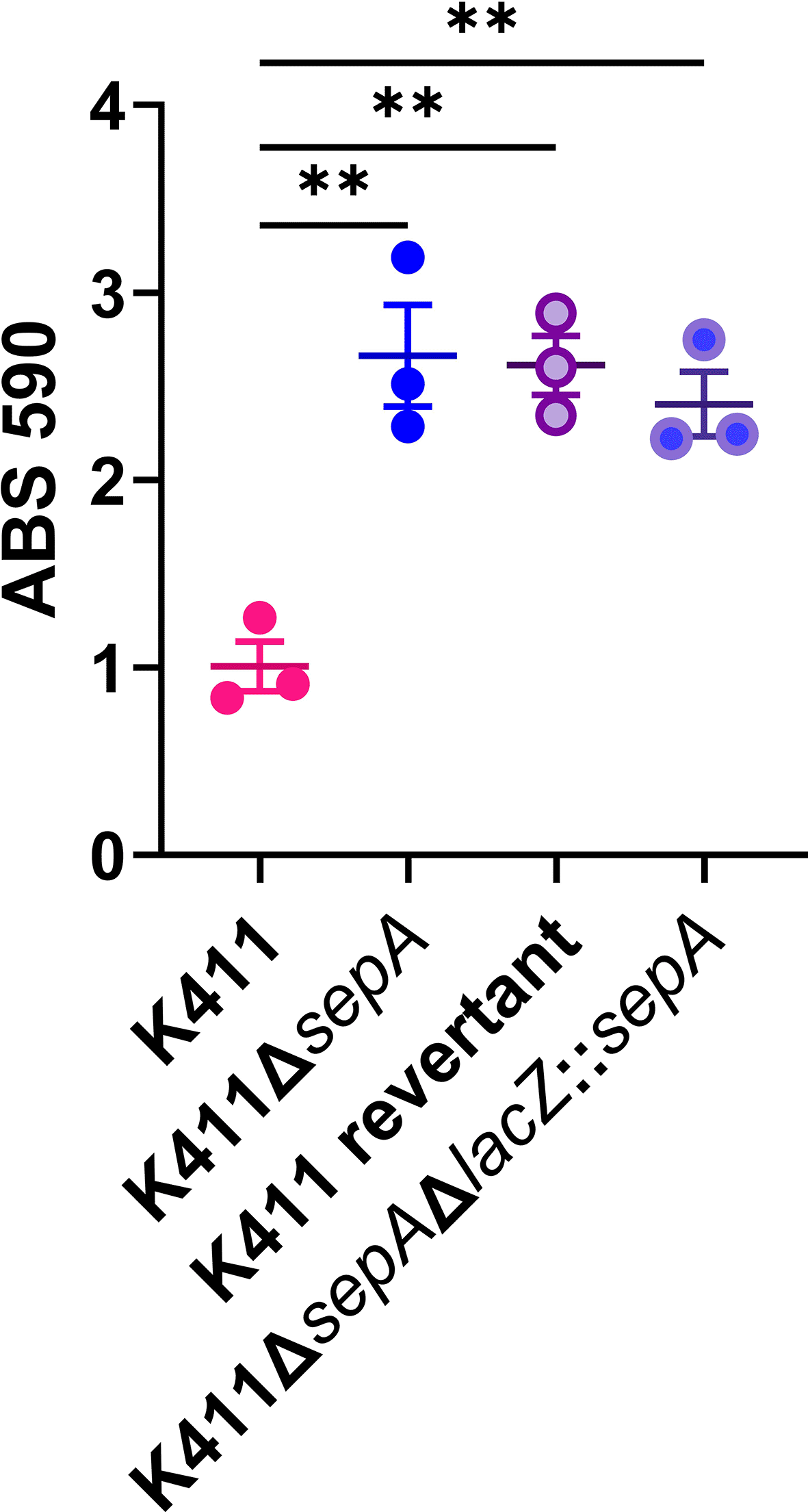
Biofilm results from a five hour 96-well plate assay. Each symbol represents the mean of four technical replicates. Error bars indicate SEM. Significance was tested by 1-way ANOVA with Šídák correction. **P≤0.01.
To assess the contribution of the SPATE SepA to the virulence of EAEC, we deleted sepA from a set of six clinical EAEC strains and assessed biofilm formation. We were surprised to find that four of the ΔsepA strains demonstrated increased in vitro biofilm staining compared with the wt controls. We hypothesized that the serine protease activity of SepA was cleaving either an EAEC surface protein or a biofilm component in the wt strains, such that when sepA was deleted that component was no longer cleaved, and biofilm staining increased. Ultimately, we did not find a direct connection between biofilm staining and SepA because we were unable to complement the mutation. However, we did find that the ΔsepA EAEC were spread further out from each other than wt EAEC in images of biofilms. This latter finding was true both for ΔsepA strains in which we observed elevated biofilm staining and for those for which we did not find elevated biofilm staining. Taken together these findings suggest that deletion of sepA does alter the biofilm to a degree, but that the effect of SepA is best observed microscopically, and is likely indirect.
We did find that pAAΔsepA conferred biofilm-forming capacity to a commensal E. coli. This result shows that increased biofilm staining is mediated solely by pAAΔsepA. We also confirmed that ΔsepA::cat alone could not confer biofilm-forming capacity to HSNalR. Taken together our results suggest for the phenotype of elevated biofilm staining the pAAΔsepA is necessary and sufficient.
As we investigated the effect of the sepA mutation, we found the following: a) all wt strains secreted similar amounts of SepA, b) all wt and ΔsepA strains had similar biofilm CFU counts, c) targeting of eDNA, protein, and carbohydrate did not alter biofilm staining levels for K261, d) deletion of the pAA tra region, as well as umuC-yccA, finO, lpxM, parB did not impact biofilm staining, and, e) increased biofilm staining for K261ΔsepA was dependent on the ability to form a biofilm.
During this investigation, we tried to complement the ΔsepA-enhanced biofilm-staining phenotype through both exogenously added protein and by genetic means. Neither crude SepA preparations or purified SepA were able to reduce biofilm staining in the mutant strain background in most cases. We did observe, curiously, that the concentrated supernatant of K411 wt decreased biofilm staining. However, neither cis- nor trans-addition of sepA reduced biofilm staining in the mutant strain background back to wt levels.
Because we ultimately realized that the increased biofilm staining found for ΔsepA EAEC strains is not due to the lack of SepA, we hypothesize that the increased biofilm staining is due to a change in the expression of other genes involved in biofilm formation on the pAA. These genes could include those for AAF expression, dispersin or dispersin transport, yet unidentified biofilm genes, or other genes involved in fimbrial gene expression, such as aggR. Another regulatory factor for EAEC is Aar, which is a repressor of aggR expression and also encoded on the pAA plasmid. However, the presence or absence of aar did not correlate with increased biofilm staining in ΔsepA strains in this study. Alternatively, it is possible that the cat insertion into sepA caused an unintended effect on the downstream AAF operon or on expression of other pAA genes. However, because sepA is located on the pAA separated from other genes by insertion elements, we do not think that deletion of sepA perturbs the expression of other genes directly.
Our data suggest that there is more work to be done on the regulation of EAEC biofilm formation/genes and the ways in which the pAA contributes to the biofilm.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)