Keywords
Antiphospholipid syndrome; Acquired hemophilia, immunosupressive therapy
Antiphospholipid syndrome; Acquired hemophilia, immunosupressive therapy
Acquired hemophilia, a rare condition, is characterized by the presence of neutralizing autoantibodies against most often Factor VIII (FVIII) and in rare cases, Factor VII (FVII). These antibodies effectively inhibit the activated form of FVII by binding to its light chain. This binding disrupts the interaction between FVIIa and tissue factor (TF). The exact mechanism responsible for the development of this condition is poorly understood. Some researchers propose a paraneoplastic context, while others suggest a link to infections and autoimmune diseases, particularly Antiphospholipid syndrome (APS).
In this case, we present a new instance of an inhibitor to FVII in a young woman experiencing a severe hemorrhagic syndrome. Notably, the symptoms were exacerbated by the administration of recombinant factor VII, and the patient had a history of APS.
In October 2022, a previously healthy 28-year-old woman presented with sudden left elbow hemarthrosis and spontaneous gingival bleeding. She had no prior personal or family history of abnormal bleeding. Upon admission, her hemoglobin concentration and platelet count were normal, but her prothrombin titer (PT) was low (11%, normal range: 70-100%), and her INR was high (7.1, normal range: 0.8-1.2). The baseline factor VII level was 6%, while the levels of other factors were normal. The patient received 5 mg of recombinant factor VII (rFVII) and was discharged the next day.
Three weeks later, the patient returned to the emergency department experiencing dyspnea and dysphagia for two days. Physical examination revealed a right cervical hematoma. A cervical computerized tomography (CT) scan without contrast showed bleeding in the retropharyngeal space, causing compression of the airways and digestive tract. Her hemoglobin concentration was 10 g/dL, and PT was still at 11%. She was admitted to the hospital and received 5 mg of rFVII. The following day, the size of the cervical hematoma increased and became bilateral. The patient developed hemoptoic sputum and ecchymosis on the right arm. PT decreased to 6%. The treatment with rFVII was resumed at a higher dose (15 mg per kg every 6 hours), and two units of red blood cells were transfused. However, the hemorrhagic syndrome worsened the next day, with heavy metrorrhagia and multiple ecchymosis on both upper and lower limbs.
A second CT scan revealed an extension of the retropharyngeal hematoma into the posterior mediastinum and retroperitoneal area, along with pericardial and pleural effusion. Treatment with tranexamic acid (1 g every 6 hours) and blood transfusion were initiated. The PT was undetectable this time, indicating the presence of an inhibitor to factor VII. The antibody titer was quantified as 428 Bethesda units (BU). Lupus anticoagulant was also detected, but other Antiphospholipid syndrome (APS) antibodies and antinuclear antibodies (ANA) were negative. The patient started treatment with methylprednisolone (1 g per day) and mycophenolate mofetil (3 g per day). Bleeding stopped within 24 hours, but the PT remained undetectable.
A week later, the patient developed deep vein thrombosis (DVT) in the left lower limb, and a bilateral segmental pulmonary embolism was incidentally discovered during a follow-up CT scan. Enoxaparin (2000 IU every 12 hours) was initiated for anticoagulation. One month after admission, the patient experienced heavy metrorrhagia again, and her hemoglobin concentration dropped to 8 g/dL. The FVII antibody titer increased to 542 IUB. Anticoagulation was discontinued, and the patient continued taking prednisone (1 mg per kg per day) and mycophenolate mofetil (3 g per day). Rituximab infusions were administered (1 g on day 1 and 1 g on day 14), which stabilized the genital bleeding. The PT increased to 8%, and the FVII antibody titer decreased to 415 BU (Figure 1).
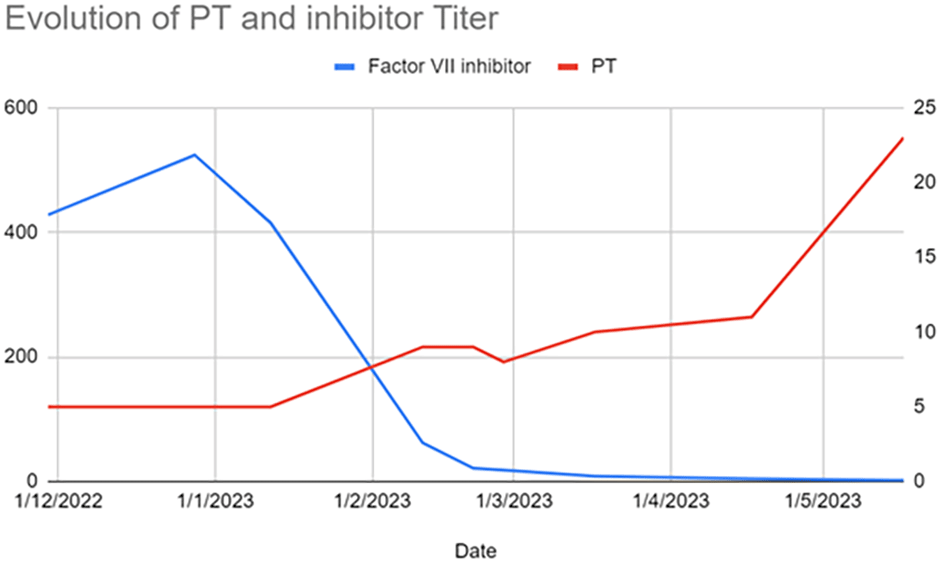
Two weeks later, the patient experienced heavy menstruation with a decreased hemoglobin concentration of 7 g/dL. Plasmapheresis was attempted, but only one session was successful, as the patient developed local catheter thrombosis during subsequent sessions. Intravenous Immunoglobulin (IVIg) therapy (0.4 g per kg per day) was administered for four days, leading to the cessation of bleeding by the third day. PT increased to 9%, and FVII antibody titer decreased to 62 BU. The patient’s prednisone dose was gradually reduced by 5 mg per week. Antibody titers continued to decrease to 23.5 and then 18.2 BU. A control test after two and a half months showed the presence of lupus anticoagulant.
After a 90-day hospitalization, the patient was discharged in April 2023. She was prescribed a daily dose of 15 mg of prednisone and 2 g per day of mycophenolate mofetil. No anticoagulation therapy was recommended. Since her discharge, she has not experienced any bleeding. Her menstruations have resumed with a normal flow and duration. Her hemoglobin concentration was 10 g/dL, PT was at 23%, and the FVII antibody titer was 2 BU.
Acquired inhibitors of coagulation are rare but severe conditions. Among them, inhibitors to factor VIII (acquired hemophilia A) are the most common, and management recommendations have been established. On the other hand, factor VII inhibitors are rarer and less well-known. These autoantibodies can be isolated or associated with various diseases. The first case report of factor VII deficiency due to the presence of its inhibitor dates back to 1980, as a paraneoplastic syndrome associated with bronchogenic carcinoma.1 Few other similar cases associated with malignancies have been reported afterward. This condition has also been found in the context of infections, mostly HIV-related.2 It has also been described in association with autoimmune diseases, mainly APS.3 Additionally, it has been reported as secondary to drug administration, particularly penicillins.4 Therefore, when encountering acquired hemophilia, thorough screening for these associated diseases is necessary before concluding its idiopathic or isolated nature.
Clinical presentations of factor VII inhibitor cases seem to be correlated with antibody titers. Severe hemorrhagic syndromes and mortality have been observed with titers above 5 Bethesda units (BU).5 To our knowledge, the titer in our report is the highest reported so far. However, the presence of neutralizing antibodies has been noted in some populations, such as patients with APS, without any clinical manifestation, making it uncertain to establish a clinico-biological correlation.6
Unfortunately, treatment strategies and objectives have not been clearly defined yet. For the symptomatic management of acute bleeding, procoagulant agents like tranexamic acid have been used in some cases.7 In our case, the use of tranexamic acid was not possible due to the patient’s thrombotic complications related to APS. Another particularity of our case is the possible role of recombinant factor VIIa (rFVIIa) in maintaining and stimulating the autoimmune process leading to the synthesis of these antibodies. Although not used in previous cases, rFVIIa has been suggested as a possible option for severe cases.8
For therapeutic options, the most common first-line choice has been a combination of glucocorticoids (GC) and immunosuppressive agents, with successful results in some cases.9 The choice of the immunosuppressive agent should take into consideration associated comorbidities to target both if possible. Cyclophosphamide and rituximab (RTX) have been commonly used. RTX has been employed for both remission induction and maintenance.10 To the best of our knowledge, this is the first reported case of a factor VII inhibitor treated with mycophenolate mofetil (MMF).11 For severe acute hemorrhages and refractory forms, intravenous immunoglobulin (IVIg) has been used with good outcomes.12 Lastly, plasmapheresis can be a potential alternative for life-threatening presentations. However, it is preferably performed before the administration of immunosuppressive drugs or IVIg to avoid increasing their clearance and thereby diminishing their pharmacological action.13
In this case report, we describe a successful treatment regimen for a severe and biologically challenging presentation of acquired factor VII inhibitor. Our approach relied on a combination of conventional immunosuppression, immunotherapy, intravenous immunoglobulin (IVIg), and plasmapheresis (PE). By employing this comprehensive treatment strategy, we effectively managed the patient’s condition and achieved positive outcomes. This highlights the importance of a multidimensional approach in the management of severe acquired factor VII inhibitors, considering both the clinical and biological aspects of the condition.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)