Keywords
Microbiota, differential abundance analysis, visualization
Microbiota, differential abundance analysis, visualization
Microbiota communities consisting of diverse microbial members, including bacteria, viruses, fungi, and other microorganisms (Berg et al., 2020) in the digestive tract, skin, respiratory and urogenital systems, and other locations (Kennedy and Chang, 2020), have emerged as a crucial factor in maintaining health and preventing disease (Hou et al., 2022). With advancements in sequencing technologies (Satam et al., 2023) there is an increasing amount of data available on the composition and function of microbiota communities (Hasan and Yang, 2019), but analyzing and visualizing these data remain challenging due to their complexity and high dimensionality (Panek et al., 2018).
Microbiota communities are organized into multiple phylogenetic levels, including Phylum, Family, Genus, and Species, with each level providing unique insights into the community’s structure and functional potential (Jandhyala et al., 2015). Integrating visualization techniques to simultaneously represent relative abundances across multiple taxonomic levels would enhance the accessibility and ease of interpretation.
Stacked bar-plot representations of the relative abundance of microorganisms remain one of the most commonly used visualizations to show the global composition of microbiota communities, as well as potential shifts in a given community (Liu et al., 2021; McMurdie and Holmes, 2013). However, current tools used to generate stacked bar visualizations in microbiota analysis do not capture phylogenetic relationships between microbial taxa (Peeters et al., 2021). This is because traditional stacked bar visualizations utilize random colors to represent different microbes within the same taxonomic level. Furthermore, differential abundance analysis, a key step in interpreting statistically significant compositional shifts and their biological implications, is typically conducted and shown separately.
To address these issues, we developed a novel R package that allows users to easily generate stacked bar-plots to visualize microbiota composition at a user-defined taxonomic level while integrating information on taxa phylogeny through the use of color gradients. In addition, statistically significant differences in relative abundance are indicated on the plot. These simple solutions allow for more information to be communicated within one single graphical representation.
StackbarExtended is implemented in native R, required version (≥ 4.0), and is platform independent, with its source code available on GitHub and archived on Zenodo (Cuisiniere, 2024a).
StackbarExtended is designed for microbiota data analysis and visualization. It requires three main inputs: a taxonomic table providing phylogenetic information, a count table detailing the abundance of the taxa, and metadata containing biological or experimental group data (Navgire et al., 2022). These tables can be obtained through established pipelines such as DADA2 (Callahan et al., 2016), Mothur (Schloss et al., 2009), and QIIME2 (Bolyen et al., 2019) in conjunction with reference databases such as Greengenes (DeSantis et al., 2006) or SILVA (Quast et al., 2013). The package supports a phyloseq S4 class object as an input, promoting a data management approach that regroups the count, taxonomy, and metadata tables into a single entity, thus simplifying data handling and enhancing reproducibility. The Phyloseq S4 object can be obtained through the phyloseq() function of the phyloseq R package (McMurdie and Holmes, 2013).
One of the StackbarExtended functionalities is the ability to handle multiple taxonomic levels simultaneously, typically focusing on user-defined levels set by default to Family (X) and Phylum (X+1). This is achieved through the use of the tax_glom() function from the phyloseq package. Most importantly, StackbarExtended uses a color-coding mechanism where taxa at a specified level (X) are visually differentiated using user-defined color palettes that match those of their Phylum level (X+1), making visual identification of the taxonomic hierarchies straightforward. This is achieved by applying different shades of the same color to represent individual taxa (X) within the same Phylum (X+1), creating a gradient effect that maps the phylogenetic taxonomy.
Another challenge in analyzing microbiota data is the large number of taxa present in the dataset. In microbiota communities, a small number of taxa typically dominate, while numerous others are present at significantly lower abundance levels (Neu et al., 2021). In classic stacked bar-plot representations such as the ones included in Phyloseq (McMurdie and Holmes, 2013), MicrobiomeAnalyst (Chong et al., 2020) or MicroEco (Liu et al., 2021) pipelines, these low-abundance taxa are simply filtered out before plotting. StackbarExtended allows users to classify taxa based on their relative abundance, and, by default, the package plots only taxa representing more than 1% of the total abundance. In addition, low-abundance taxa (X) can be grouped into their respective “Others” taxonomic level and the information about their taxonomy (i.e. Phylum) is kept while taxa belonging to low abundance Phyla (X+1) are grouped into a general “Others” category. Thus, by still including the low-abundance taxa, StackbarExtended provides a more accurate representation of the taxa in a microbiota community.
Finally, StackbarExtended includes DESeq2 differential abundance analysis functionality (Love et al., 2014) which allows users to statistically assess the difference in taxa abundance between experimental groups and apply fdr corrections. Significant features (i.e. taxa with significant differences in abundance between 2 groups) are highlighted in the legend using bold font, and the significance levels (p-value) after fdr corrections are automatically shown using stars in the legend. When more than two treatment groups are compared, multiple pairwise comparisons can be computed and data-frames are produced with results providing information about the taxa identified as significant at each phylogenetic level analyzed through the DESeq2 analysis. This information includes the taxa names and phylogeny, their corresponding abundance levels, statistical metrics such as log2 fold-change, p-values and fdr-corrected p-values.
To demonstrate the use of this package, we have provided ready-to-use example data “ps” from our previously published study (Cuisiniere, 2024a; Cuisiniere et al., 2021) which assessed shifts in mouse gut microbiota composition after antibiotic treatment (Figure 1).
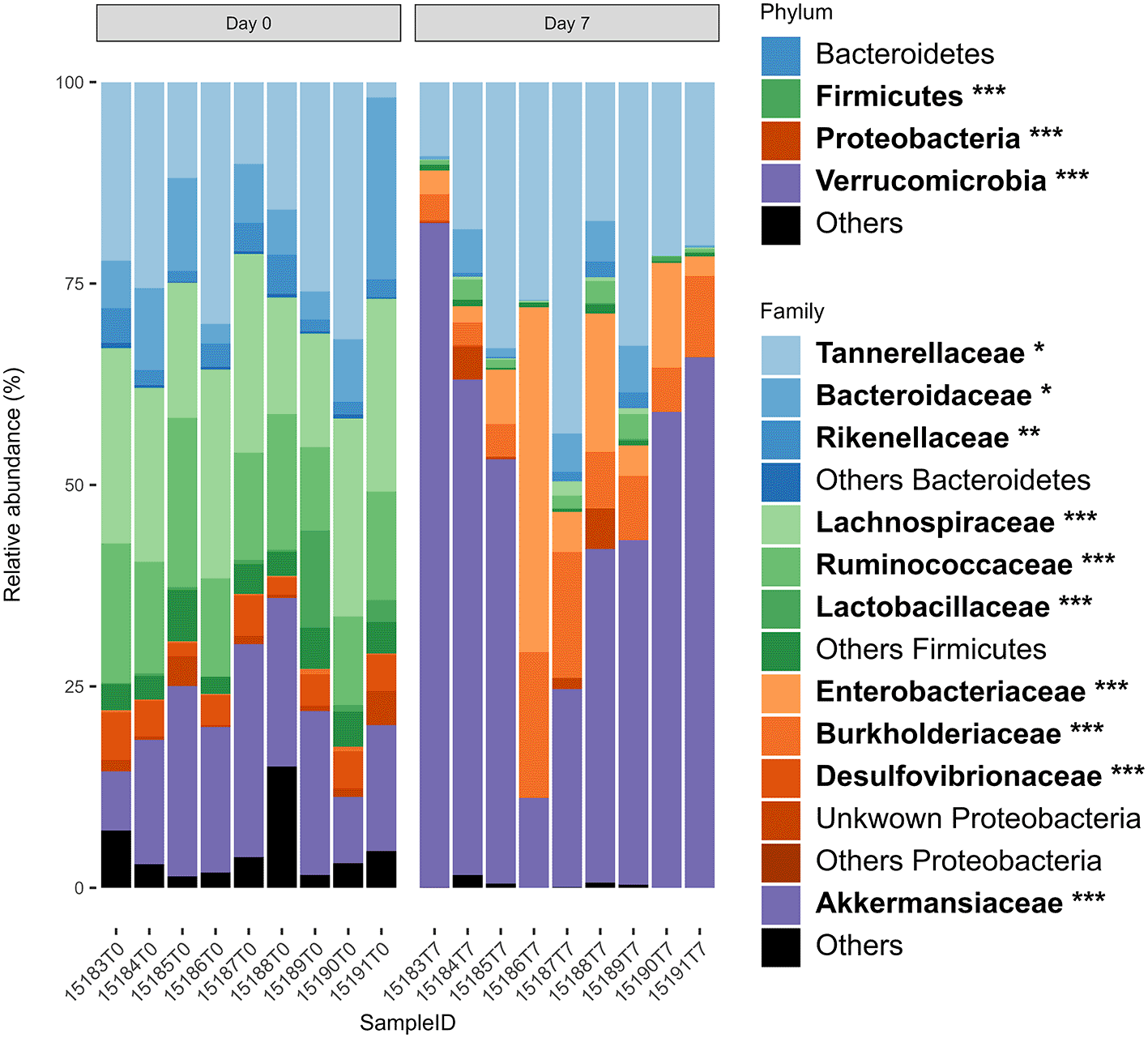
Mice (n = 9) received oral antibiotic treatment with neomycin and metronidazole for one week. Fecal samples were collected before (Day 0) and after antibiotic treatment (Day 7). 16S rRNA of DNA extracted from fecal samples was sequenced using the Illumina MiSeq platform. The 4 most abundant phyla and families are represented. Families with a mean abundance lower than 1% across the samples are regrouped into “Others”. Taxa represented in bold within the legends are statistically significant after fdr correction (*P < 0.05, **P < 0.01, ***P < 0.001). Data used are from the “ps” dataset and are included in the StackbarExtended R package.
Animal experiments were approved by the Institutional Animal Care committee of the Centre de recherche du Centre hospitalier de l’Université de Montréal (CRCHUM) in agreement with the guidelines of the Canadian Council of Animal Care. The study was carried out in compliance with the ARRIVE guidelines (Cuisiniere, 2024b). No criteria were set for including and excluding animals during the experiment or data points during the analysis. No exclusions of animals, experimental units, or data points were applied for the analysis.
In order to obtain the data, four-week-old female C57Bl/6 mice were purchased from Charles River Laboratories (Saint-Constant, QC, Canada). Constant efforts were made to minimize the suffering of the animals. Nine mice were kept under controlled specific pathogen free (SPF) conditions in the CRCHUM animal facility at a temperature of 22°C, 45-60% humidity with a light-dark cycle of 12-12. They were housed at three mice per cage with ad libitum access to chow and water. Cages were enriched with nesting material. Mice were allowed one week of acclimation following arrival to the CRCHUM animal facility, after which oral antibiotics consisting of metronidazole (1 mg.ml−1, Hospira, St-Laurent, QC, Canada) and neomycin (1 mg.ml−1, Sigma, St-Louis, MO, USA) were added to the drinking water for one week. Fecal samples were collected before (Day 0) and after antibiotic treatment (Day 7), snap-frozen and stored at -80°C. Mice were then euthanized using CO2 followed by cervical dislocation. DNA was extracted using the Qiagen DNeasy PowerSoil® kit (Qiagen, Toronto, ON) and quantified using a spectrophotometer (DeNovix DS-11 FX, Wilmington, DE). The 16S ribosomal RNA (rRNA) library preparation and sequencing was performed using the Illumina MiSeq platform at Genome Québec targeting the V3-V4 (Primers: 341F, 805R) region of the 16S rRNA gene. Forward and reverse, raw, demultiplexed 16S rRNA reads were denoised, chimera filtered, and clustered into sequence variants using the Dada2 package (version 1.16) (Callahan et al., 2016) in R (version 4.0.1). Reads were trimmed at the first instance of a quality score less than or equal to 2 or removed if they contained ambiguous nucleotides (N) or if two or more errors were expected based on the quality of the trimmed read. After taxonomic assignment using Silva training set v132 (Quast et al., 2013), ASV (Table 1), taxonomy (Table 2) and metadata (Table 3) were combined into a phyloseq object (McMurdie and Holmes, 2013).
Rows represent each sample and columns represent individual ASVs. Each cell indicates the count of a particular ASV in a specific sample.
| ASV1 | ASV2 | ASV3 | ASV4 | ASV5 | |
|---|---|---|---|---|---|
| 15186T0 | 2035 | 2717 | 160 | 851 | 346 |
| 15189T7 | 4119 | 2552 | 3 | 41 | 209 |
| 15187T0 | 2757 | 868 | 552 | 15 | 389 |
| 15190T7 | 4173 | 3 | 0 | 27 | 1 |
| 15188T0 | 2535 | 1230 | 87 | 445 | 434 |
Rows represent individual ASVs, and columns represent each taxonomic rank. Each cell contains the taxonomic name at that rank for the corresponding ASV.
Rows contain metadata associated with each sample.
Users can then use the following code to create graphical representation of the gut microbiota composition at the phylum and family levels comparing mice before and after antibiotic treatment and performing differential abundance with fdr correction.
# The plot and the output tables are stored into a single list object
my_plot <- plot_microbiota(
ps_object = ps,
exp_group = 'timepoint',
sample_name = 'SampleID',
hues = c("Purples", "Blues", "Greens", "Oranges"),
differential_analysis = T,
sig_lab = T,
fdr_threshold = 0.05
)
print (my_plot$plot)
In addition to the graphical representation, two output tables (one for each level) are created containing statistical information concerning the differentially abundant taxa (Table 4). The tables can be accessed as follows:
#Display statistically significant taxa at the phylum level print(my_plot$significant_table_main) #Display statistically significant taxa at the family level print(my_plot$significant_table_sub)
The data frame output contains the results columns: baseMean, log2FoldChange, lfcSE, stat, pvalue (unajusted p-values) and padj (fdr-corrected p-values), and also includes metadata columns of related taxonomic information. The lfcSE gives the standard error of the log2FoldChange. For the Wald test, stat is the Wald statistic: the log2FoldChange divided by lfcSE, which is compared to a standard Normal distribution to generate a two-tailed pvalue. For the likelihood ratio test (LRT), stat is the difference in deviance between the reduced model and the full model, which is compared to a chi-squared distribution to generate a p-value.
The primary objective of our example dataset is to identify which taxa at the Family and Phylum levels were impacted by antibiotic treatment in mice gut microbiota. This analysis is crucial for understanding how antibiotic interventions alter microbial communities. The graphical output (Figure 1) provided a clear assessment, revealing that among the four most abundant Phyla, three were significantly affected by the antibiotic treatment. Specifically, significant alterations were observed in Verrucomicrobia, Bacteroidetes, and Proteobacteria (fdr < 0.05), indicating a substantial shift in the microbial composition due to antibiotic exposure. Among the abundant Families (>1% of the total abundance) within these Phyla, 10 out of 11 exhibited significant alterations (fdr < 0.05). On average, the six remaining low-abundance Phyla, grouped into the “Others” category, accounted for 2.5% of the total relative abundance. Similarly, within the abundant Phyla, the 14 low-abundance Families accounted for 2.4% of the total relative abundance.
The two generated output tables provide detailed statistical information about the differentially abundant taxa at each level, including log2 fold-change, exact p-values, and fdr-corrected p-values. This comprehensive data allows for identification of taxa significantly impacted by the treatment. Notably, four additional low-abundance Phyla - Cyanobacteria, Deferribacteres, Tenericutes, and Actinobacteria - were found to be significantly affected (fdr < 0.05). Furthermore, a total of 24 families were identified as statistically different (fdr < 0.05) (Cuisiniere, 2024a).
These findings are consistent with previous studies that have reported significant shifts in gut microbiota composition following antibiotic treatment (Fishbein et al., 2023). Hence, this use case demonstrated the advantage of using StackbarExtended to present a clear and interpretable graphical representation while retaining the capacity to perform detailed statistical analysis.
StackbarExtended offers the opportunity to enhance the widely utilized stacked bar graphical representation by incorporating information about taxonomy and statistical significance in regard to differentially abundant taxa. Furthermore, it enables users to retain data on rare taxa while maintaining phylogeny information. These functionalities have been implemented into a user-friendly R package, StackbarExtended, which is freely accessible on GitHub. The package facilitates the processing of large microbiota datasets and produces publication-ready and information-rich graphical representations with a high level of personalization. StackbarExtended is particularly useful to biology and molecular biology students, fellows and researchers working in microbiota analysis, and its output visualizations are suitable for publications and time-limited presentations at conferences and seminars requiring quick interpretation of displayed data.
Source code available from: https://github.com/ThibaultCuisiniere/StackbarExtended.
Archived software available from: https://doi.org/10.5281/zenodo.11166800 (Cuisiniere, 2024a).
License: This R package and underlying data are freely available under the Gnu Public License (GPL-3).
Animal experiments were approved on April 3rd 2019 by the Institutional Animal Care committee of the Centre de recherche du Centre hospitalier de l’Université de Montréal (CRCHUM) in agreement with the guidelines of the Canadian Council of Animal Care, approval number C19006MSs. The study was carried out in compliance with the ARRIVE guidelines (Cuisiniere, 2024b).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)