Keywords
Oseltamivir, Hydroxamic acid, Molecular hybridization, Anti-influenza agents, Neuraminidase.
Oseltamivir, Hydroxamic acid, Molecular hybridization, Anti-influenza agents, Neuraminidase.
Influenza viruses exert a significant threat to public health and are highly associated with variability and recombination.1–3 Certain known strains of the influenza virus usually mutate, and the genomic segments may undergo reassortment to produce new virus subtypes.4,5 Owing to the variable biochemical nature of viruses, the production of vaccines and synthesis of potential drugs is facing great challenges. Nevertheless, continuous potential research has led to the production of two main types of agents for the clinical treatment of influenza virus, such as, neuraminidase inhibitors (NAIs) and M2 channel ion blockers.6,7 NA inhibitors can weaken the release of influenza virus particles from infected cells, and effectively inhibit viral replication. The most potent representatives of NA inhibitors and FDA-Approved drugs include; Oseltamivir, Peramivir, and Zanamivir.8,9 Oseltamivir has been widely used in the treatment of influenza viruses and is a competitive inhibitor of NA.10,11 However, owing to the evolution of the influenza virus and the abuse of anti-influenza drugs, drug-resistant strains have emerged, such as H274Y/H1N1, which are the culprit leading to the H1N1 pandemic.11–13 Besides, the side effects of oseltamivir, such as inhibiting the production of viral antigens, leading to the reduction of acquired antiviral humoral immunity, and increasing the probability of re-infection have been recorded.14 Oseltamivir is a bulky molecule that cannot fit the active site of neuraminidase alone. However, it can bind to cavities 150 and 430, which induces a conformational change in neuraminidase, exposing the active site and allowing the oseltamivir molecule to bind to the active site and inhibit neuraminidase.15 NAIs that target cavities focus on designing molecules that can bind to both cavities with high affinity. This is a challenging task, as the two cavities are relatively small and hydrophobic.16 Several NAIs that target the cavities have been developed, and a number of the most promising candidates including Peramivir, Laninamivir octanoate, and Baloxavir marboxil are resistant to mutation.17,18 Certain amino acids have the potential to be employed as agents to facilitate targeted drug penetration and distribution. The solubility, stability and cellular penetration of antiviral agents can be enhanced through conjugation with amino acids, thereby, enhancing their delivery to infected cells.16 The enhanced biocompatibility of hybrid antiviral agents containing amino acids contributes to improved safety profile.19 The introduction of an amino acid substituent to the C1-carboxyl group of oseltamivir via an amide bond to afford carboxamides displayed greater potency.17 Histone deacetylases (HDACs) facilitate influenza virus replication by promoting chromatin condensation. The potential hindrance of viral genome replication can be achieved through inhibition of HDACs activity.13 Hydroxamate-based neuraminidase inhibitors function by structural mimicry, wherein they adopt configuration similar to that of sialic acid. The binding of a hydroxamate moiety to neuraminidase obstructs the active site of the enzyme, thereby, impeding its ability to cleave sialic acid residues. This mechanism inhibits the detachment of the virus from infected cells and impedes its propagation to adjacent cells.13,14 These inhibitors were designed to possess enhanced potency, a wider spectrum of activity, and reduced incidence of adverse effects.20 The molecular hybridization approach has been employed in the advancement of novel pharmaceuticals with enhanced efficacy, an expanded range of effectiveness and diminished adverse reactions.21
In view of previous investigations, an approach for synthesizing oseltamivir carboxamides with amino acids that may show better anti-influenza activity and probably enhance physicochemical properties is considered. L-serine and L-isoleucine, L-tryptophan and L-tyrosine are linked via amide bonds with oseltamivir carboxyl group. These carboxamides were also subjected to further reactions using hydroxylamine to afford the hydroxamates as molecular hybrids. These hydroxamates are expected to exert synergistic effects and, consequently, enhance the anti-influenza activity of oseltamivir.
Oseltamivir phosphate (CAS No. 196618-13-0), L-tyrosine (CAS No. 60-18-4), L-isoleucine (CAS No. 73-32-5), L-serine (CAS No. 56-45-1), L-phenylalanine (CAS No. 63-91-2) and hydroxylamine hydrochloride (CAS No. 5470-11-1) were purchased from Sigma/Aldrich (St. Louis, MO, USA). Triethylamine (CAS No. 121-44-8) and dimethylsulphoxide (DMSO, CAS No. 67-68-5) were obtained from BDH (India). MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Influenza Neuraminidase Inhibitor Susceptibility Assay Kit (ab283398/abcam/USA). http://www.abcam.com/ab283398.
The melting points were determined using an electrical melting point apparatus (Electro-thermal 9300, USA). Infrared spectra were recorded on a (KBr disk) using an FT-IR spectrophotometer (Shimadzu 8400). 1H-NMR spectra were recorded using a Bruker 500 MHz spectrophotometer and 13C-NMR spectra (Avance III, 75.65 MHz spectrophotometer) were recorded using dimethylsulphoxide (DMSO-d6) as the solvent (5 mL). Chemical shifts were recorded in parts per million (ppm). Tetramethylsilane (TMS) was used as an internal reference. In vitro cytotoxicity assay was performed using the MTT colorimetric assay. The MTT assay is the first rapid colorimetric assay developed for cell viability with high screening in a 96-well format. This assay measures the reduction of yellow MTT to an insoluble blue formazan product by mitochondrial succinate. Neuraminidase inhibitor susceptibility tests were conducted using the Influenza Neuraminidase Inhibitor Susceptibility Assay Kit to evaluate the activities of the investigated compounds.
ADME parameters and drug-likeness were determined using Swiss ADME (server). Lipinski’s rule of five holds substantial importance in the preclinical stage of pharmaceutical development and was used to predict the molecular properties of the investigated compounds.
The chemical structures of the compounds were depicted using ChemDraw Professional software (version 19.0) and energy minimization was performed using the MM2 force field. The structure of the neuraminidase enzyme was retrieved from the Protein Data Bank (PDB code: 3CL0).
Molecular docking was performed using Hermes software (2021.2.0), an integral part of the CCDC GOLD suite. The ligand selected for the docking procedure included all protein residues situated within a 10 Å radius of the binding site on the protein. The determination of a fitness score is based on docking performance as assessed by the GOLD algorithm. The level of fitness is positively correlated with the strength of the docking interaction between the protein and ligand. Discovery Studio (2021) was used to create a 3D view (H-bonding cavity) and 2D view of the selected results (poses) of the docking process. The docking scores of the oseltamivir carboxamides and their hydroxamate molecules were recorded.
A mixture of an amino acid (0.01 mole, L-phenyl alanine, L-tyrosine, L-serine or L-isoleucine) and oseltamivir phosphate (0.01 mole) in absolute ethanol (30 mL) containing sodium hydroxide (5%) was continuously stirred and refluxed for 7 h. The mixture was cooled, poured into cold water (100 mL) and neutralized with diluted hydrochloric acid to pH 6. The solid product was collected and crystallized in ethanol: water (3:7). Chemical structures of the synthesized compounds were confirmed by spectral analyses (FT-IR, 1H-NMR and 13C-NMR). Details are provided in data availability.
Oseltamivir-Phenylalanine (2-({[4-(acetylamino)-5-amino-3-(pentane-2-aryloxy) cyclohex-1-en-1-yl] carbonyl} amino)-3-phenylpropanoic acid) was synthesized using oseltamivir phosphate (0.01 mole, 4.08g) and L-phenylalanine (0.01 mole, 1.65g), as previously described.
Oseltamivir-Isoleucine (2-(4-acetamido-5-amino-3-(pentan-3-yl) oxy) cyclohex-1-ene-1-carboxamido)-3-methylpentanoic acid) was synthesized using oseltamivir phosphate (0.01 mole, 4.08g) and L-Isoleucine (0.01 mole, 1.17g), as previously described.
Oseltamivir-Serine (2-({[4-(acetylamino)-5-amino-3-(pentane-2-aryloxy) cyclohex-1-en-1-yl] carbonyl} amino)-3-propanoic acid) was synthesized using oseltamivir phosphate (0.01 mole, 4.08g) and L-serine (0.01 mole, 1.05g), as previously described.
Oseltamivir-Tyrosine (2-({[4-(acetylamino)-5-amino-3-(pentan-2-yloxy) cyclohex-1-en-1-yl] carbonyl} amino)-3-(4-hydroxyphenyl) propanoic acid) was synthesized using oseltamivir phosphate (0.01 mole, 4.08g) and L-tyrosine (0.01 mole, 1.81g), as previously described.
A mixture of oseltamivir carboxamides (1 mM) and zinc dust (0.5 g) in dioxane (30 mL), triethylamine (1 mM) and hydroxylamine hydrochloride (1 mM) was stirred continuously and refluxed for 2 h. The reaction mixture was filtered and carefully poured into cold water (100 mL) with continuous stirring. A crystalline powder was formed, separated by filtration and recrystallized from dioxane: water (3:7). Chemical structures were confirmed by spectral analyses (FT-IR, 1H-NMR and 13C-NMR). Details are provided in data availability.
Oseltamivir-Phenylalanine (1 mM, 0.431 g) and zinc dust (0.5 g) in dioxane (30 mL) containing TEA (1 mM, 1.2 mL) and hydroxylamine hydrochloride (1 mM, 0.069 g) were treated as previously described to obtain Os-Phe-hydroxamate.
Oseltamivir-Isoleucine (1 mM, 0.383 g) and zinc dust (0.5 g) in dioxane (30 mL) containing TEA (1 mM, 1.2 mL) hydroxylamine hydrochloride (1 mM, 0.069 g) and treated as previously described to afford Os-Ile-hydroxamate.
Oseltamivir-Serine (1 mM, 0.385 g) and zinc dust (0.5 g) in dioxane (30 mL) containing TEA (1 mM, 1.2 mL) and hydroxylamine hydrochloride (1 mM, 0.069 g) were continuously stirred and treated as previously described to afford Os-Ser-hydroxamate.
Oseltamivir-Tyrosine (1 mM, 0.447 g) and zinc dust (0.5 g) in dioxane (30 mL) containing TEA (1 mM, 1.2 mL) and hydroxylamine hydrochloride (1 mM, 0.069 g) mixture were treated as previously described to afford Os-Tyr-hydroxamate.
The cytotoxicity assay was performed using the MTT colorimetric assay22 and the effects of the synthesized compounds were evaluated in the MDCK cell lines.
MDCK cells were purchased from the American Type Culture Collection (ATCC) and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM). Media were supplemented with heated fetal bovine serum (10%), L-glutamine (1%, 2 mM) and penicillin (100 IU/mL)/streptomycin (100 μg/ml) from (Euro Clone). The cell growth profile was seeded in a 96-well plate at a density of 6×104 cells/well and cell viability was determined by trypan blue exclusion using a haemocytometer.
MDCK Cells were washed with phosphate-buffered saline (PBS) and detached with 0.025% trypsin-EDTA (Euro Clone), and media was added to a volume of 10 ml. The cell suspension was centrifuged at 1000 rpm for 10 min, and the pellets were re-suspended in 10 ml medium to make a single cell suspension. Cell viability was determined by trypan blue exclusion, which exceeded 90% as counted by a haemocytometer. The cells suspension was diluted to obtain the optimal seeding density, and 100 μl aliquot was placed in a 96-well plate. The cells were cultured at 37°C in a humidified atmosphere of 5% CO2, incubated for 24 h and then treated with different concentrations of each compound starting from 10mM (dissolved in DMSO, 5 mL) in a serial dilution manner (10, 5, 2.5, 1.25, 0.62, 0.31, 0.15 and 0.078 mM) and further incubated for 72h to determine the IC50 values. Cell growth was analyzed using the MTT colorimetric assay at the end of the exposure time.
The newly synthesized compounds were tested for the cell viability using an MTT colorimetric assay.22 MTT stock solution (15 μl, 5 mg/ml) in sterile PBS (pH 7.4) was incubated for 72 h and placed in each well. The cells were then added to each well and incubated for a further 3 h in the presence of a solubilizing stop solution (100 μL) to solubilize the dark violet formazan crystals. The optical density was measured at 570 nm using a microplate reader. The results are expressed as the percentage of cell viability compared with the control, corresponding to untreated cells.
The IC50 values for the investigated compounds were determined by constructing a dose-response curve.23 In the MTT assay, IC50 values denote the concentrations of compounds necessary to achieve a 50% reduction in cell viability. Based on the acquired data, the IC50 values were determined after a 72 h of exposure of the cells to the tested compounds. To ascertain the IC50 values, a concentration range consisting of the following concentrations (10-0.078 μM) was used.
Determination of the appropriate dilution factor for H1N1 viral strain
4-Methylumbeliferone (4-MU) was used as a standard and by diluting the stock solution (5 mM, 10 μL) to neuraminidase assay buffer (990 μL) to obtain the working solution (50 μM). Using the compounds at concentrations (0, 0.1, 0.2, 0.4, 0.8, 1.2, 1.6 and 2.0 nM/well) and the 4-MU standard was prepared by adding 0, 2, 4, 8, 16, 24, 32 and 40 μL of the 50 μM solution into a series of wells, and the volume was adjusted for each well to 100 μL with neuraminidase assay buffer. Neuraminidase Stop Solution (100 μL) was added to each well (final volume of 200 μL/well) and mixed thoroughly. The fluorescence of the solution in the wells was measured at Ex/Em = 368/460 nm, the 0 nM/well (RFU) reading was subtracted from all of the standard readings, and the slope of the 4-MU standard curve was constructed. A series of 12 reaction wells from the H1N1 viral strain (one row of a 96-blanck well plate) was prepared by adding neuraminidase assay buffer (50 μL) to each well in columns 1-12. Fifty microliters of undiluted H1N1 viral isolate were added to column 1 and mixed thoroughly. A series of 2-fold serial dilutions were performed across the row of wells by transferring 50 μL from column 1 to column 2 and mixing the contents, until column 11 was reached. Fifty microliters from column 11 were discarded, leaving column 12 to serve as a background control well (no virus) to correct for non-enzymatic substrate hydrolysis. The volume of each well was 50 μl. The (96-dark well plate) was incubated at 37°C for 10 min to equilibrate the contents of the wells to the reaction temperature. During incubation, a concentrated neuraminidase substrate working solution (2X) was prepared by diluting the reconstituted neuraminidase substrate stock solution (100X) with neuraminidase assay buffer at a 1:50 ratio. Neuraminidase substrate solution (50 μl, 2X) was added to each reaction well, and the volume was brought to 100 μL/well. The plates were incubated at 37°C for 60 min in the dark. The reaction was terminated by addition of Neuraminidase Stop Solution (100 μL) to each well. The contents were thoroughly mixed and the fluorescence of the solutions was measured at Ex/Em= 368/460 nm.
Determination of the dilution factor corresponds to the RFU value
The fluorescence (F) was quantified by subtracting the mean fluorescence intensity of the solution containing no virus background control wells (RFU blank) from the fluorescence intensity of each sample well (RFU sample): F = RFU sample – RFU blank.
For the H1N1 viral strain, background-subtracted F values were plotted against the sample dilution factor. This value was used as the viral dilution factor in the inhibitor susceptibility assay.
A set protocol was used to evaluate NA susceptibility, as follows; each tested compound was dissolved in DMSO solvent at final concentrations of less than (2%, v/v) to produce a master stock solution (10 mM). Working solution (1 mM) was prepared by diluting the master stock with neuraminidase assay buffer at a 1:10 ratio. The working solutions (4X) were prepared in a range of concentrations by diluting the working solution in neuraminidase assay buffer (to generate a multi-point dose-response curve) and to determine IC50 values for each compound. A series of working solutions (ten, 4X) were prepared in neuraminidase assay buffer (0.04, 0.2, 0.4, 2, 4, 20, 40, 400, 4000, and 40000 nM, corresponding to a final concentration range of 1 μM to 10 μM). H1N1 viral strain to be tested for susceptibility, a series of reaction wells were prepared containing 25 μL of each 4X test concentration (working solution) as well as the corresponding no-inhibitor control (containing neuraminidase assay buffer, 25 μL) and background control wells (containing 50 μL neuraminidase assay buffer). The H1N1 viral isolate stock solution was diluted with neuraminidase assay buffer according to the optimal dilution factor determined in the neuraminidase activity viral titration assay. The plate was incubated at 37°C for 30 min to allow the inhibitors to interact with the viral enzymes. During the incubation, a concentrated neuraminidase substrate working solution (2X) was prepared by diluting the reconstituted neuraminidase substrate stock solution (100X) with Neuraminidase assay buffer at a 1:50 ratio. A neuraminidase substrate solution (50 μL, 2X) was prepared for each well.24
The reaction was started by adding a neuraminidase substrate working solution (50 μL, 2X) to each reaction well, bringing the volume to 100 μl/well. The plates were incubated at 37°C for 60 min in the dark. The reaction was terminated by adding a neuraminidase stop solution (100 μL) to each well. The contents were mixed thoroughly and the fluorescence of all wells was measured at Ex/Em = 368/460 nm. For each reaction well, including no inhibitor/vehicle controls, the fluorescence intensity of the background control well (RFU blank) was subtracted to determine the background-corrected fluorescence (denoted by F). For each NI test concentration (FNI) in the dose-response curve, the percent inhibition (or remaining activity) relative to the vehicle control (FVC) was calculated using the following equation:
The relative activity (or percent inhibition) at each NI concentration was plotted and the IC50 values were calculated by non-linear logistic curve fitting.
All oseltamivir carboxamides with amino acids showed higher PLP fitness than that of oseltamivir acid. Os-Phe had the highest PLP fitness (72.228), while, Os-Ile had the lowest PLP fitness (59.142), when compared with oseltamivir acid (PLP fitness 56.241). The docking scores are listed in Table 1. The basic chemical structures of the target oseltamivir carboxamides include the potential pharmacophore of an oseltamivir ring containing a free carboxyl group, which is essential for its activity. Therefore, the interaction of these target compounds and oseltamivir with the amino acids of the enzyme is expected to be the same, as illustrated in Table 1, which is why the docking scores or PLP fitness scores showed great similarity.
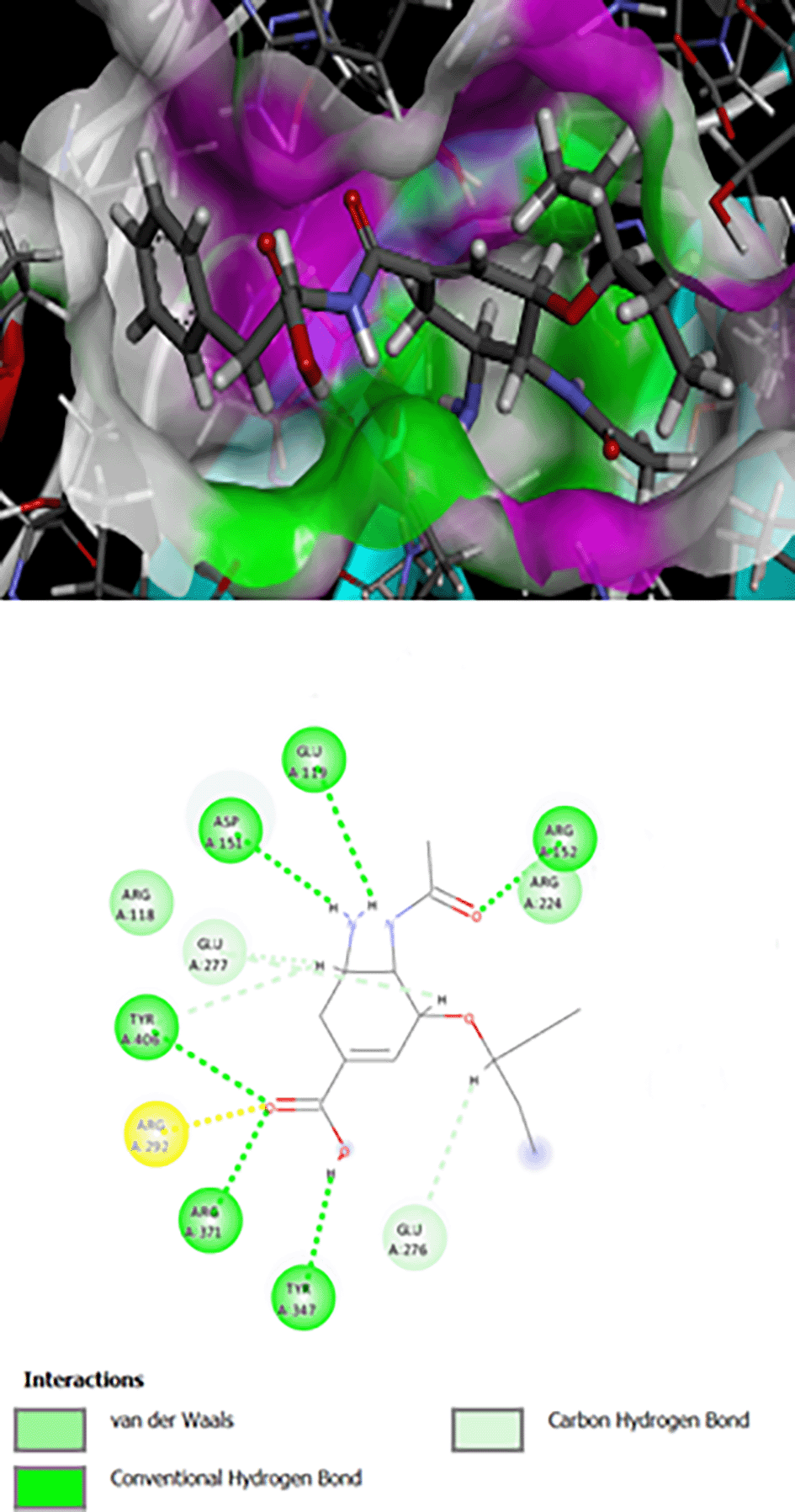
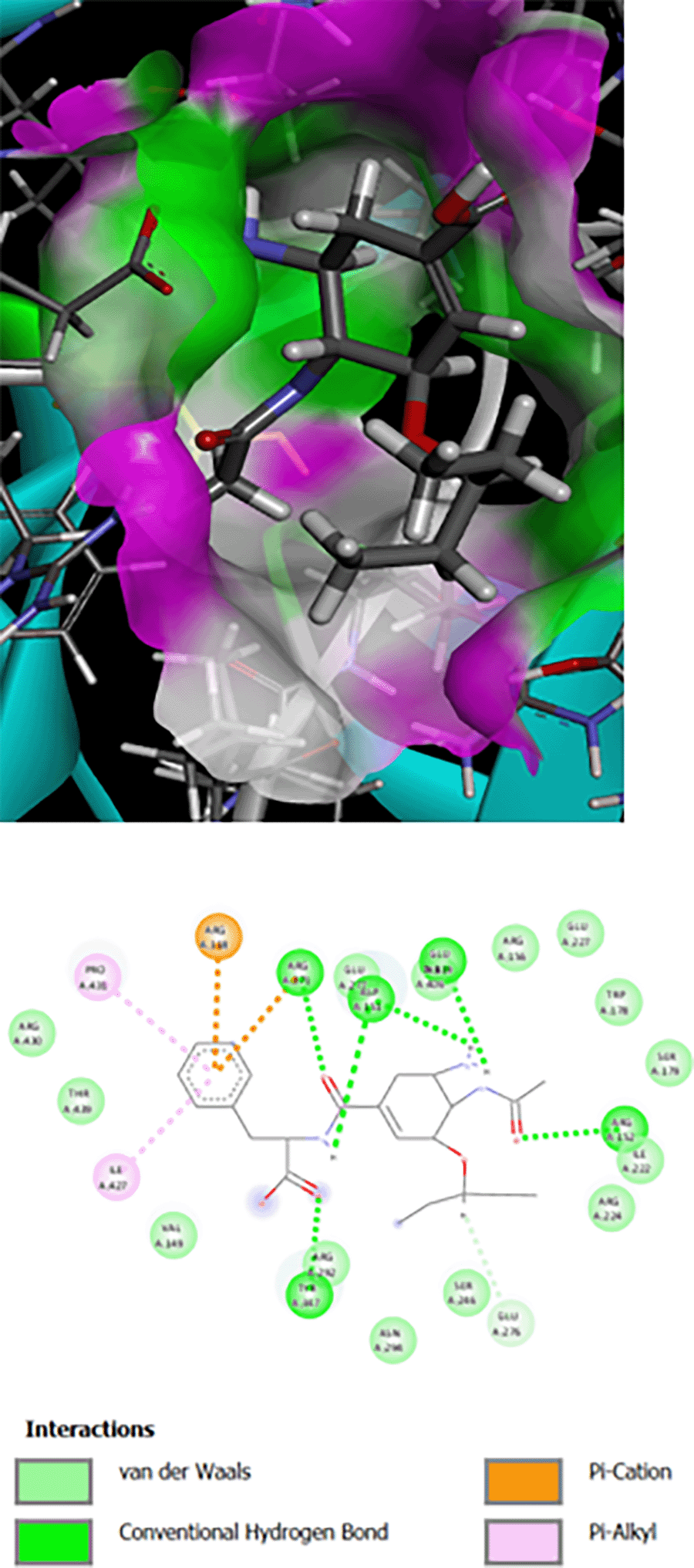
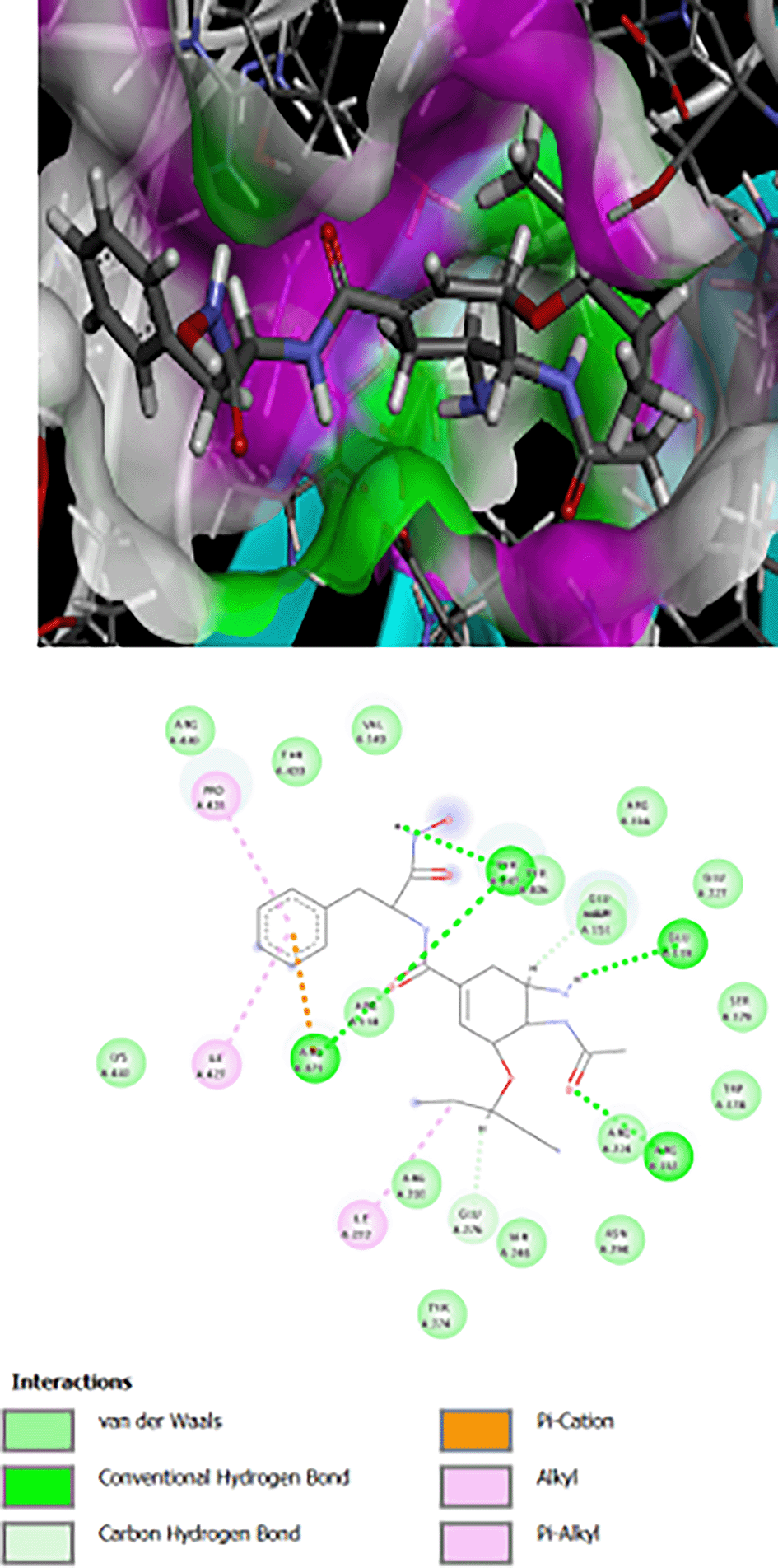
The interaction of Os-Phe and its hybrid with hydroxamic acid and oseltamivir on N1 influenza neuraminidase enzyme type 3CLO is presented as 2D and 3D binding maps (Figures 1, 2, 3). The amino acids involved in the interaction of these compounds with neuraminidase indicated somewhat strong binding, as the binding was with several amino acids (Table 1). All compounds have the same binding requirements to reflect their activities and show higher affinity to the target protein. The 3D binding maps show the ability of the compounds to bind to cavities 150 and 430, while oseltamivir binding is restricted to cavity 150, which may reflect the higher PLP fitness of the synthesized target compounds compared to oseltamivir. It was obvious that the interaction of oseltamivir and its carboxamides and the hybrid molecules with hydroxamic acid occurred with the very basic amino acid, arginine at various positions (Table 1). This is probably due to the presence of the free carboxyl group of the oseltamivir ring, the amide in its carboxamides and the hydroxamate groups in the hybrid molecules. All the above groups are acidic in nature and interact with the basic arginine residues of the enzyme.
The molecular properties of the investigated target compounds were calculated based on Lipinski’s rule and its components.25 Lipophilicity (miLogP) and the total polar surface area (TPSA) values are essential factors for predicting the oral bioavailability of drugs.26 TPSA is a very useful descriptor used to characterize drug absorption and bioavailability, permeability through Caco-2 cells and transport across blood brain barrier.26 Higher values for TPSA and OH-NH interactions indicate that these compounds may have smooth and efficient binding to receptors. The recorded TPSA for all synthesized target compounds were higher than that of oseltamivir, indicating the possibility of potent binding affinity to the target enzyme. This may explain the higher docking scores obtained for oseltamivir acid (Tables 1 and 2). However, drug molecules with TPSA values of 140 Å or higher are expected to exhibit very low absorption.27 The ADME parameters of the new oseltamivir carboxamides and their hybrid molecules were measured to predict the possibility of oral absorption and reveal the safety of selecting the potential candidate(s). Os-Phe and Os-Ile have TPSA values of 132.75 and were predicted to have good permeability and GI absorption. Compounds Os-Phe, Os-Phe-hydroxamate, Os-Tyr and Os-Ile and oseltamivir acid showed higher lipophilicity values, which may be due to the hydrophobic groups present in their chemical structures (Table 2). All the synthesized target compounds showed no violation of the Lipinski rule of five, except for compounds Os-Ser-hydroxamate and Os-Tyr-hydroxamate, which may be due to the presence of hydroxyl groups at both amino acid side chains in serine and tyrosine. These violations were recorded for the H-donor and H-acceptor values. The BOILED-EGG, a graphical representation of all the calculations performed in order to forecast two crucial ADME parameters; passive absorption from the gastrointestinal tract (GIT) and access to the brain across the blood-brain barrier (details are found in data availability). The target compounds had a bioavailability score of 0.55, indicating that all compounds may enter the systemic circulation, as presented for Os-Phe and its hydroxamate derivative. Oseltamivir and the investigated compounds showed no penetration through the BBB, and may be considered Pgp substrates. The investigated compounds and oseltamivir were not Cytochrome P enzymes inhibitors, as tested for the following types Cyp1A2, Cyp2C19, Cyp2C9, Cyp2D6 and Cyp3A4.
The synthetic procedures for the target compounds is illustrated in Scheme 1. The basic principle of these methods is the synthesis of oseltamivir carboxamides with certain amino acids using a simple and effective procedure with appreciably high yields, which is an ester aminolysis reaction, as previously described. The most acceptable mechanism for these reactions is shown in Scheme 1. Oseltamivir carboxamides are converted to hybrid molecules by further reaction with hydroxylamine to afford oseltamivir-amino acid hydroxamates. The characterization and physical properties of the target compounds are presented in Table 3.
The proposed chemical structures of the synthesized compounds were confirmed by spectral analyses (FT-IR, 1H-NMR, 13C-NMR), as illustrated in Table 4 (details are listed in data availability).
The half maximal inhibitory concentrations (IC50 μM) of the investigated compounds on MDCK cells were compared to those of oseltamivir and the findings are presented in Table 5.
All oseltamivir carboxamides showed less cytotoxic activity and an excellent safety margin (Table 5). However, hybrid molecules of oseltamivir-amino acids-hydroxamates showed higher cytotoxic activity than oseltamivir (1490.5). This is an expected result, as the linkage of amino acids to certain drugs has been shown to afford much better conjugates in terms of activity, membrane penetrations, and enhanced physicochemical properties.17,19
Statistical calculations of the results were conducted using Microsoft Office-Excel, and Figure 4 depicts the dose-response curves for IC50 values of the investigated compounds.
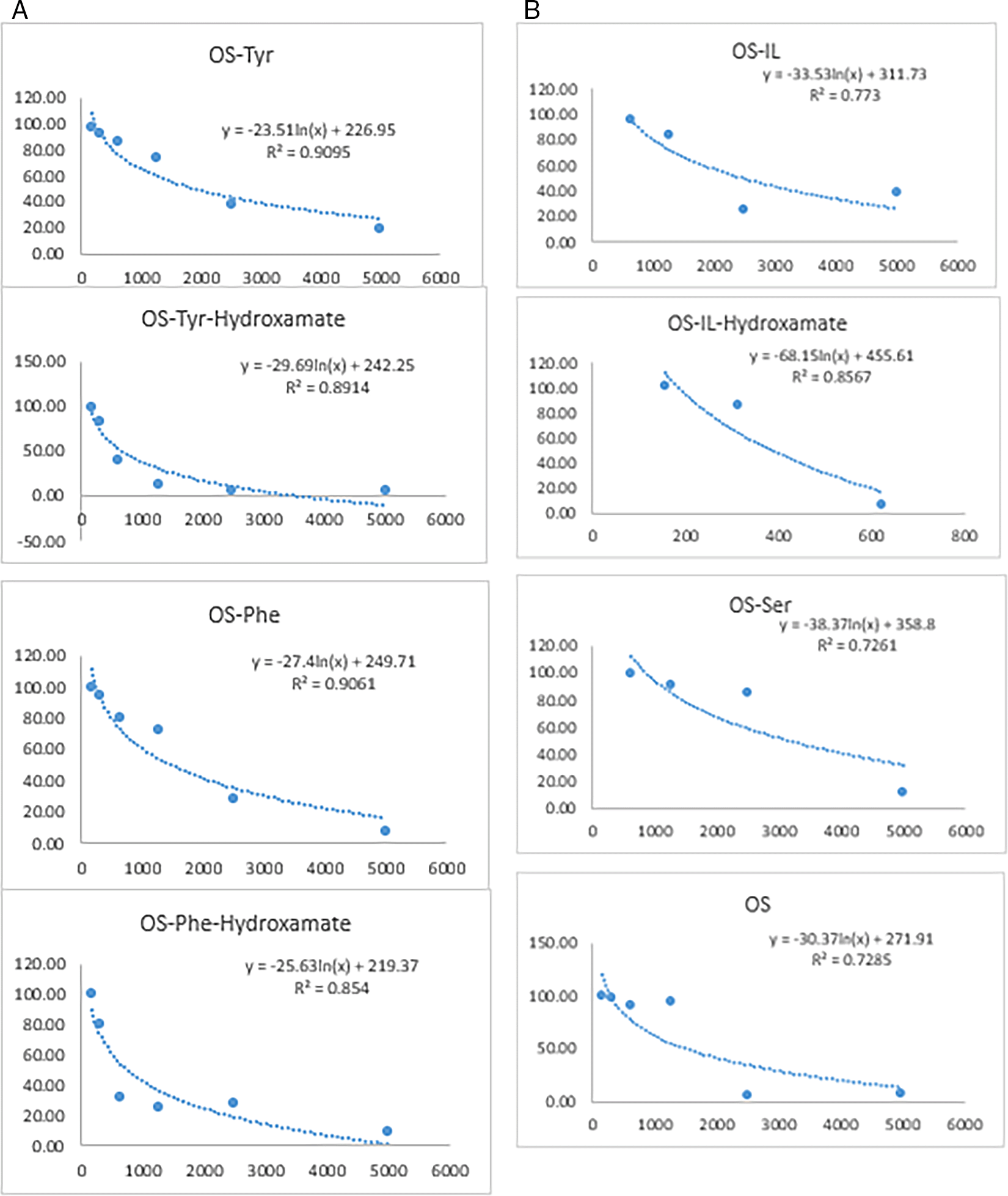
The in vitro cytotoxic activity against MDCK cell lines was used to determine the comparative cell viability percentage in relation to oseltamivir over 72 h of incubation. The percentage viability of oseltamivir carboxamides was comparable to that of oseltamivir, indicating great similarity with an acceptable safety margin. However, oseltamivir carboxamide-hydroxamates showed a much lower percentages of viable cells. At high concentrations (5000, 2500, and 1250 μg/ml) oseltamivir and its carboxamides with L-tyrosine, L-phenylalanine, L-isoleucine and L-serine had moderately high percentages of viability (72.45-94.04 μg/ml) indicating their moderate safety margin. Moreover, at a lower concentration of 156 μg/ml of all the investigated compounds including, oseltamivir, the percentage of viability was comparable and recorded very high percentages approaching 98% and above (details are listed on data availability).
The in vitro evaluation of anti-influenza activity, represented as the percent inhibition (or remaining activity) of neuraminidase at each concentration, was plotted to calculate the IC50 values by non-linear logistic curve fitting (Figure 4). The results revealed that there was a great variation in the activity of the investigated compounds of series one and two. Os-Phe exhibited the highest and most remarkable activity. This may be due to the presence of an aromatic ring of phenylalanine on the side chain, which may have contributed to the binding affinity, and consequently, enhanced the activity. However, Os-Tyr did not show appreciable activity, which may be due to the presence of a phenolic hydroxyl group that may have shifted the binding of the ligand away from the active site of the enzyme. Moreover, Os-Ile showed very low inhibitory activity against neuraminidase (5060 μM), while, Os-Ile-hydroxamate showed much better activity (216.4 μM), although, it was still less than that of oseltamivir. Os-Phe showed better anti-influenza activity (3.03 μM) than oseltamivir activity (67.22 μM). However, the hybrid molecule, which is Os-Phe-Hydroxamate recorded an activity approaching 430 μM, which is less active than Os-Phe (3.03 μM) and oseltamivir (67.22 μM). Oseltamivir-amino acid-hydroxamates were less active than oseltamivir carboxamides or oseltamivir. This may indicate that these hybrid molecules have activity on HDACs enzymes and not on the neuraminidase of influenza. This case definitely requires further investigation to evaluate their activities on HDACs, as inhibitors. This possibility cannot be ruled out. It is quite obvious that aliphatic amino acids did not contribute to activity, and hence, the IC50 values of Os-Ile, Os-Ser, and their hydroxamates were very poor and recorded much higher IC50 values. Similar results were obtained for oseltamivir carboxamides with alanine and glycine.28 Interestingly, oseltamivir carboxamide with D-valine, exerted high potency against neuraminidase of H5N1 and H5N6. The inhibitory potency of NA may be ascribed to the retention of the carboxylate group and the presence of a hydrophobic isopropyl group. However, the incorporation of alanine or glycine did not improve activity.28 Similarly, in the present study, oseltamivir carboxamides with L-serine and L-isoleucine did not show appreciable activity for not a clear reason, when compared with the active oseltamivir carboxamide with D-valine, as mentioned earlier. Apparently, the presence of a free carboxyl group or its isostere, such as an amide, on the ring structure of oseltamivir is important for the inhibition of neuraminidase to afford an anti-influenza activity. A similar finding of improved activity was established for oseltamivir carboxamides with various amines.28 The active site or loops of neuraminidase are large enough to accommodate an inhibitor with reasonable activity.15 Amino acids possessing aromatic or cationic side chains have the ability to engage in interactions with viral proteins or nucleic acids, so impeding viral reproduction.29 Incorporation of aromatic amino acids within certain cephalosporin have proved effective in enhancing the binding affinity of the parent molecules leading to an increase in activity.30 Synthesis of oseltamivir carboxamides has proved to be effective and retain activity and in certain cases, such as, aromatic amino acids enhanced the activity over the parent compound. This has also been observed in synthesizing new levofloxacin carboxamides with certain aromatic amino acids.31 Hydroxamate-derived compounds have demonstrated efficacy as neuraminidase inhibitors, rendering them presently employed in the treatment and prophylaxis of influenza infection.32 Oseltamivir-hydroxamates, containing a CONHOH group instead of a COOH group, exhibited a significant decrease in activity (> 40-fold) compared to the parent compound. The CONHOH group was not suitable for accommodating NA pocket. This pocket in the active site contains three basic residues, Arg 118, Arg 292, and Arg 371, which can strongly interact with acidic groups, such as a COOH32 or the phosphoryl group.33 It is not surprising that the above oseltamivir carboxamides32 revealed much weaker activity and this may be due to the lack of the linker side chain, and hence, does not fulfil the required structure-activity relationship (SAR) for the optimum activity. The required length is 4-6 atoms separating the cap group and the hydroxamate moiety. The proposed oseltamivir carboxamides containing aliphatic and aromatic amino acids in the side chain included in this study retain the full SAR requirement for optimum activity. Generally, the hydroxamate group, with an intramolecular hydrogen bond is weaker than COOH in acidity, which may have largely contributed to the poor activities of these compounds.
Two series of novel oseltamivir carboxamides and their hybrid molecules with hydroxamic acid were successfully synthesized and evaluated using in silico methods. The results showed that all compounds were highly absorbed via passive diffusion through the gastrointestinal tract and comply with Lipinski’s rule of five, except Os-Ser-Hydroxamate and Os-Tyr-hydroxamate with one violation for each. The in vitro evaluation of anti-influenza activity revealed that there was a great variation in the activity of the investigated compounds in both series one and two. Os-Phe exhibited the highest and most remarkable activity. Generally, oseltamivir-amino acids-hydroxamates are less active than oseltamivir carboxamides or oseltamivir.
Alwan, S.M.: Project idea, supervision, data curation, formal analysis, acquisition, methodology and chemical synthesis, resources, visualization, writing – original draft preparation, review and editing. Tayah, S.S.: Formal analysis, methodology and chemical synthesis, in silico prediction, investigation, resources, validation, writing draft preparation.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)