Keywords
Myositis, Polymyositis, Dermatomyositis, Anti-synthetase Syndrome, Antibodies, Diffuse Interstitial Pneumonia
Myositis, Polymyositis, Dermatomyositis, Anti-synthetase Syndrome, Antibodies, Diffuse Interstitial Pneumonia
After considering the comments and suggestions from the first reviewer, we have decided to submit a revised version of the article. In this revision, we have aimed to provide a more detailed explanation of the methods section.
Similarly, we have added a figure to better illustrate the distribution of the population, highlighting the use of two inclusion criteria and the overlap of certain patients.
We have also revised the abstract for a better summary of this purely descriptive study, as we took into account that some results in the previous abstract were not included in the study.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Idiopathic inflammatory myopathies (IIM) encompass a group of rare acquired autoimmune diseases, primarily characterized by muscular inflammation accompanied by varying degrees of motor weakness.1 Despite this shared foundation, IIM exhibit significant heterogeneity both clinically and histologically. Indeed, within IIM, various muscular phenotypes are observed, where muscular weakness can be limited or absent, reflecting different mechanisms of muscular damage.2 Furthermore, IIM can be associated with extra-muscular manifestations, including lung, heart, skin, gastrointestinal tract, and joint involvement,3 or with cancer, which also reflects diverse pathophysiological mechanisms.4 This clinical polymorphism, as well as the rarity of these pathologies, are challenging for physicians trying to identify homogeneous patient subgroups.5 This diversity is reflected in the multiple proposed classification criteria developed since 1975. At that time, the first attempt to categorize IIM was published by Peter and Bohan, dividing them into two historical subgroups: polymyositis (PM) and dermatomyositis (DM).6 Over time, the spectrum of these diseases has expanded from conditions primarily characterized by myositis to true systemic diseases.7 Simultaneously, the deep understanding of the pathophysiological mechanisms of IIM and notably the discovery of myositis specific antibodies (MSAs) has contributed to identify new IIM entities, such as immune-mediated necrotizing myopathy (IMNM) and anti-synthetase syndrome (ASS). Consequently, this original classification has come under scrutiny,8 and subsequent classifications have been reshaped, with growing number of published criteria for the classification and/or diagnosis of IIM, including criteria for PM, DM, ASS and IMNM, though not completely able to conceal the heterogeneity within this group.7 The scientific literature continuously advances in understanding these diseases, particularly regarding the role of MSAs and their relevance for patient classification.1 Nevertheless, few studies have focused on IIM in Tunisia, especially considering these recent developments. Hence, we conducted this study aiming to describe the characteristics of IIM in Tunisian patients.
We conducted a retrospective study including patients diagnosed with IIM from December 2001 to August 2022 in the internal medicine department of the Rabta Hospital in Tunisia.
Included patients had to fulfill the 2017 American college of rheumatology/European league against rheumatism (ACR/EULAR) classification criteria for IIM (meaning patients who had the necessary minimum probability (55%) to be classified as having IIM9).
Patients who met the ASS criteria, proposed by Connors et al,10 were also included in the study. Patients with juvenile dermatomyositis and inclusion body myositis (IBM) were excluded. Patients with IIM according to the ACR/EULAR classification were divided into three subgroups based on the classification tree: DM, PM, and amyopathic dermatomyositis (ADM) and patients who met the criteria of Connors et al were classified into the ASS subgroup.
Demographic data, clinical findings, results of laboratory investigations, electromyography, and muscle biopsy findings, were collected from medical records, using a pre-established survey.
IIM associated with another connective tissue disease (CTD) was retained when the patient met the criteria for IIM in addition to the criteria for the CTD, namely ACR/EULAR 2013 classification criteria for systemic sclerosis, ACR/EULAR 2019 classification criteria for systemic lupus erythematosus, ACR/EULAR 2016 classification criteria for Sjögren’s syndrome and ACR/EULAR 2010 for rheumatoid arthritis.
IIM associated with cancer was considered when the patient met the criteria for IIM and was diagnosed with a histologically documented cancer.
The indirect immunofluorescence assay (IIFA) on HEp-2 cells (human cells originating from laryngeal carcinoma) was used for detecting antinuclear antibodies (ANA). ANA were considered positive when ≥ 1/80.
Patients were assessed for MSA and myositis-associated antibodies (MAA) with extractable nuclear antigens (ENA), detected using enzyme-linked immunosorbent assay (ELISA) that allowed the simultaneous detection of autoantibodies against SSA/RO52, SSA/RO60, SSB/La, SM, U1-RNP, Scl70, Jo-1, histones, and centromere, and Line blot immunoassay (EUROLINE Autoimmune Inflammatory Myopathies, EUROIMMUN), utilizing strips sensitized with recombinant peptides and purified native peptides. Results are visually evaluated based on the positivity or negativity of the antigen, with a cut-off of 1+. The panel of MSAs assessed depended on the type of Kit available in the hospital laboratory. The search for anti-HMGCR antibodies was not conducted in any patient (not available in our Laboratory).
We analyzed the data using the Statistical Package for the Social Sciences (IBM SPSS Statistics) version 2611 Categorical variables are reported as numbers (percentages) and numerical variables as either mean ± standard deviation or median and interquartile range (IQR).
The study included 97 patients. The mean age at the time of IIM diagnosis was 48.4 ± 13.8 years, with a predominance in the age group between 41 and 60 years (51%). The study population consisted in 70 females (72%) and 27 males (28%), with a male-to-female gender ratio of 0.36.
Our cohort was composed by referring to two sets of criteria, proposed for the classification of IIM and for the diagnosis of AAS, respectively.
The ACR/EULAR criteria allowed the inclusion and the classification with IIM of 85 (88%) patients (probability score ≥ 55%) (Table 1). One patient, who is labeled as having PM in our department (diagnosed in 2003 according to the Peter and Bohan criteria), had possible PM according to the ACR/EULAR criteria (ACR/EULAR probability score = 51%) and was not included.
| Definite (ACR/EULAR score≥ 90%) N (%) | Probable (50%< ACR/EULAR score ≤55%) N (%) | Total N (%) | |
|---|---|---|---|
| DM | 43 (44) | 3 (3) | 46 (47) |
| ADM | 3 (3) | 1 (1) | 4 (4) |
| PM | 12 (12) | 23(24) | 35 (36) |
| Total | 57 (59) | 27 (28) | 85 (88) |
Connors et al criteria allowed the inclusion and classification as ASS of 34% (33/97) of the patients, including those excluded by the ACR/EULAR classification (ACR/EULAR probability score < 50%) (n=12; 12%).
Noteworthy, a group of patients (n=21, 22%) fulfilled both sets of criteria, and their subgroups varied depending on the criteria used: ASS according to Connors, and DM (n=6), PM (n=14), and ADM (n=1) according to ACR/EULAR criteria.
Figure 1 and Table 2 illustrate the distribution of our study population, according to both inclusion criteria.
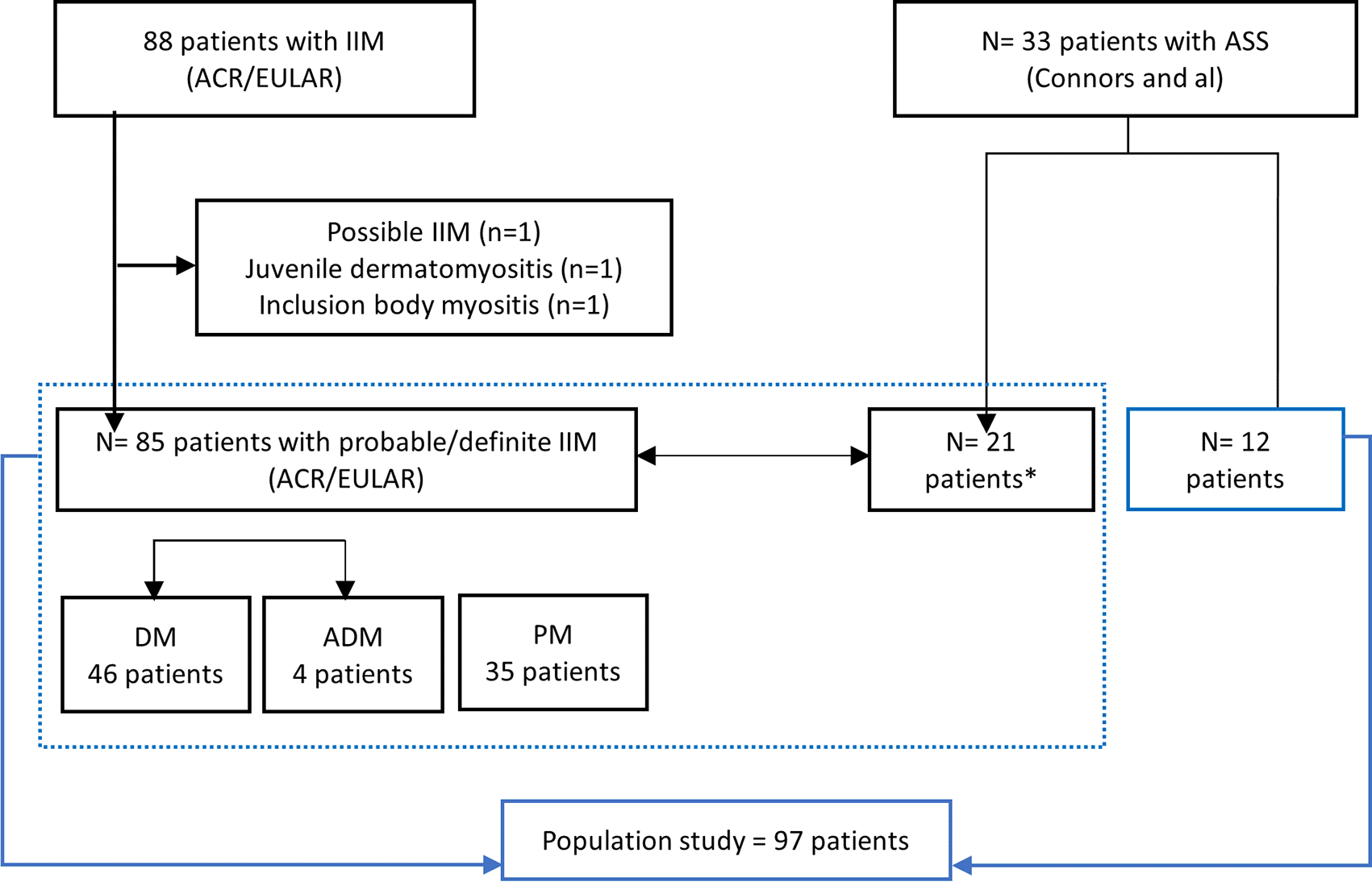
*Patients who met both criteria (Connors and ACR/EULAR).
DM: Dermatomyositis, PM: Polymyositis, ADM: Amyopathic Dermatomyositis, IIM: Idiopathic Inflammatory Myopathy, ASS: Anti-Synthetase syndrome.
Diagnostic delay and initial manifestations
The median time between symptoms onset and IIM diagnosis was five months [IQR = 2-12], with a range from two weeks to 15 years. Muscular involvement was the most frequent revealing manifestation of IIM (40%). The clinical heterogeneity of IIM was shown by the diversity of extra-muscular initial manifestations: cutaneous (32%), joint (22%), pulmonary (19%), and general signs (5%).
Muscular involvement was observed in 85 patients (88%). Amyopathic forms of IIM were exclusively reported in the ADM subgroup (2/4, 50%) and the ASS subgroup (11/33, 33%). The involvement of skeletal muscles was constant (n=85, 88%). The clinical and paraclinical features of muscular involvement are summarized in Table 3.
Cutaneous involvement
Cutaneous involvement, noted in 85% of patients, was the most common manifestation following muscle involvement, with 18 different cutaneous signs described. Skin manifestations, traditionally recognized as pathognomonic and characteristic of DM, were prevalent within the DM and ADM subgroup in our series, including heliotrope rash (88%), rash in sun-exposed areas (66%), Gottron’s papules (60%), manicure sign (32%), Gottron’s sign (24%), and psoriasiform erythematous-violaceous eruption (24%). Less frequently, other signs were described in DM and ADM patients such as cutaneous vasculitis (14%), calcinosis (10%), skin ulcers (10%), panniculitis (8%), and palmar papules (2%). Similarly, cutaneous involvement in ASS was frequent (91%) and diverse, with palmoplantar hyperkeratosis (52%) and Raynaud syndrome (36%) being predominant. Notably, pathognomonic, and characteristic DM signs were also observed among these patients: heliotrope rash (15%), Gottron’s papules (12%), periungual erythema (12%), shawl and V- sign (6%) and Gottron’s sign (3%). Nailfold capillaroscopy, performed on 21 IIM patients, was pathological in 95% of cases. Although non-specific microangiopathy was the most described aspect (13/21; 62%), scleroderma pattern defined by the presence of mega-capillaries, was observed in seven patients (33%).
Cardiac and pulmonary involvement
Cardiac involvement noted in 14 patients consisted in pericarditis (9%) and myocarditis (8%). It was diagnosed based on cardiac MRI or scintigraphy. Pulmonary involvement was found in 51% of IIM patients.
Diffuse infiltrative lung disease (ILD) was the most common pulmonary manifestation (45%). ILD was mostly common among ADM patients (4/4; 100%) and ASS patients (28/33; 85%), followed by PM (20/35; 57%) and DM (8/46;17%).
The characteristics of cardiac and pulmonary involvement are summarized in Table 4.
Joint involvement
Articular manifestations were found in 48% of IIM patients. Inflammatory arthralgias were reported in 48% of patients and synovitis in 19% of patients. Joint involvement was most commonly polyarticular (68%). Radiological bone erosions were identified in three patients (6%), two of whom had associated RA.
Serological profile
Antinuclear antibody (ANA) testing, conducted on 92 patients, was positive in 79% of cases. Anti-extractable nuclear antigen antibodies (anti-ENA), assessed in 91 patients, were found in 50% of them.
Myositis dot was performed in 54 patients: It contained 11 myositis-specific antigens (Mi2, NXP2, TIF1, MDA5, SAE, Jo1, PL7, PL12, SRP) in only eight patients, seven myositis-specific antigens (Mi2, Jo1, PL7, PL12, OJ, EJ, SRP) in 48 patients and five myositis-specific antigens (anti-Mi2, anti-Jo1, anti-PL7, anti-PL12, anti-SRP) in all the 54 patients. Since anti-Jo1 is also included in the anti-ENA panel, it was tested in 91 patients.
Myositis specific antibodies (MSA) were detected in 52% of cases, with a predominance of anti-synthetase antibodies (ASA). Among these patients, four (8%) had more than one MSA: Anti-SRP (3+) associated with anti-Mi-2 (1+) in one patient, anti-Jo-1 (1+) in one patient, and anti-PL-7 (1+) in one patient, and anti-PL-7 (3+) associated with both anti-SRP (1+) and anti-Mi-2 (1+), in the fourth patient.
MAA were found in 50 patients (52%). Patients’ immunological findings are summarized in Table 5.
Associated pathologies
IIM was associated with another CTD in 23% of patients. CTDs were: systemic sclerosis (35%), Sjögren’s syndrome (35%), systemic lupus erythematosus (21%) and rheumatoid arthritis (7%).
IIM was associated with neoplasia in 16 patients (17%), 12 of them classified as DM (12/46, 26%). Cancer diagnosis was established within three years of the IIM diagnosis in 75% of cases (12/16). It was prior to IIM diagnosis in two patients, with intervals of 1 and 5 months, respectively, concurrently during the neoplastic investigation conducted after IIM diagnosis in five patients and was made after IIM diagnosis in nine patients, with an average time interval of 39.4 months. Cancers were revealed by IIM in half of the patients, among whom seven had no initial clinical indicators. Reported neoplasia were colorectal cancer (n=4), breast cancer (n=4), lung cancer (n=2), malignant hemopathy (n=2), cervical, endometrial, nasopharyngeal and kidney cancer in one case each. Noted histological types were adenocarcinoma (n=12), squamous cell carcinoma (n=2), hepato-splenic T-cell lymphoma (n=1) and myelomonocytic leukemia (n=1).
Our cohort was composed by referring to two sets of criteria proposed for the classification of IIM and for the diagnosis of ASS, respectively. This choice was driven by the absence, to date, of classification criteria identifying all subgroups of IIM.
In line with our study, the female predominance in IIM is a consistent finding in various global studies,12 as well as in Tunisia.12 In a systematic literature review, the age at diagnosis was 55.1 ± 19.5 years, with an initial peak during childhood (juvenile DM) and a second peak in adulthood.12 In our cohort, the mean age at diagnosis was 48.4 ± 13.8 years. This difference could be explained by the exclusion of juvenile MI and IBM in our study. Given the rarity of IIM and the methodological heterogeneity evident in the various selection criteria used in studies, an accurate estimation of their frequency remains a challenge for researchers.13 The first literature review aiming to determine the global incidence and prevalence of IIM found an overall incidence of 7.98 cases per million people per year and a prevalence of 14 per 100,000 inhabitants. This review did not identify well-established geographical disparities, although the incidence of DM appeared to follow a latitudinal gradient, possibly explained by the immunomodulatory effects of ultraviolet rays.12 In Tunisia, the incidence and prevalence of IIM have not been determined, but they are among the least common systemic CTD. In accordance with our study, a significant predominance of DM compared to PM has been reported in Tunisian patients.14
The classification of IIM remains a subject of numerous discussions and debates.5 Historically, two entities were distinguished, by Peter and Bohan: DM and PM. These criteria included cutaneous and muscular signs, and by definition, the latter must be present for the classification of PM or DM.6 This straightforward classification was widely used. However, it had a major limitation: the two defined entities were heterogeneous. Initially, it did not allow the isolation of IBM.4
IBM was incorporated into the Dalakas criteria in 1991. These criteria, provided a more detailed description but were based on expert opinions.15 While some researchers focused on histological findings and underlying pathological mechanisms, others described MSAs.5 Since, many other classifications were proposed to define other entities based on the progress and the discovery of new antibodies or on immunohistochemical staining.16–19 Despite the multitude of proposed criteria, most have been based on expert opinions, primarily relying on clinical and histological data, and do not encompass all subgroups of IIM.7 To address these shortcomings, a panel of experts was convened in 2004 to develop new criteria, culminating in the long-awaited ACR/EULAR classification.9 When compared to Peter and Bohan,6 Tanimo,16 Targoff,17 Dalakas,15 and ENMC criteria,18 Targoff’s performed slightly better. No other set showed both higher sensitivity and specificity simultaneously. Furthermore, they allowed for proper classification of IIM subgroups in most cases. Thus, this classification brought significant improvements to existing criteria by proposing criteria developed from a large population of patients with and without IIM and was validated in external cohorts. The ease of use and the availability of an online calculator are also major advantages.20 However, since the development project was launched in 2004, several MSA were unknown, or their detection tests were not accessible. Consequently, several items, such as muscle MRI data and most MSA outside of anti Jo-1, were not included.9 Therefore, this classification appears clinically oriented, limiting its performance in distinguishing IIM subgroups.21 Our study illustrated these shortcomings: Twelve patients who were classified as ASS according to Connors’ criteria were not included by the ACR/EULAR classification, due to the absence or insufficient manifestation of muscle-related symptoms and the presence of other ASA apart from anti-Jo-1. Additionally, among the 21 patients classified as ASS according to Connors’ criteria and acknowledged by the ACR/EULAR classification, their distribution spanned across other subgroups. These findings highlight the limitations of the ACR/EULAR classification in accurately capturing the full spectrum of cases, especially those involving patients with non-Jo-1 antibodies and less apparent muscle involvement. On the other hand, the Connors criteria,10 adopted in our study for ASS diagnosis, are based on expert opinions and may possibly rely excessively on ASA, which can lead to over diagnosing this condition, in the absence of standardized immunological tests.22
In traditional descriptions, IIM were recognized as diseases primarily affecting muscles. Muscular involvement was the common denominator in the majority of classifications, and its absence ruled out the diagnosis4. With the identification of new subgroups and the emergence of MSAs, we have witnessed a paradigm shift in the spectrum of IIM, where muscular involvement can be limited or absent.2 It was the case of 12% of our patients, exclusively classified as ADM and AAS. This finding reflects the inclusion criteria adopted, namely the ACR/EULAR criteria that recognize ADM and Connors’ criteria that acknowledge ASS in the absence of clear muscular manifestations. Although distal muscles are traditionally spared in IIM apart from IBM,23 they were involved in 5% of our patients. A proximo-distal deficit affecting dorsiflexion of the feet and extensors of the fingers has been reported subsequently in advanced forms.24 The utility of Electromyography lies in its high sensitivity and specificity for diagnosing IIM as well as its capability to rule out differential diagnoses. In line with our study, a “myogenic” or “mixed” pattern is frequently described in electromyography recordings in IIM patients.25 Normal electromyogram tracing should not, however, rule out the diagnosis, depending on the number of explored muscles, the timing of the test, the experience of the performer, and any previous use of corticosteroids.25,26 Although IIM share histopathological features such as mononuclear cell infiltrates and areas of necrosis, certain abnormalities have been linked to distinct entities.27 In PM, muscle fiber alteration is mediated by cytotoxic cellular mechanisms. Histological examination typically reveals inflammatory cells surrounding, invading, and destroying fibers, resulting in infiltrates within fascicles. In DM, infiltrates are perivascular, in septa around fascicles, and less commonly endomysial.28 Consistent with these findings, infiltrates were predominantly perivascular and perifascicular in our DM patients, while they were predominantly intraparenchymal in our PM patients. This result is expected since the ACR/EULAR classification takes this contrast into account.9 In line with other studies, perifascicular atrophy, long considered pathognomonic for DM,29 has been described in ASS.30 Necrotic and regenerating muscle fibers were more frequent in PM patients, probably reflecting the prevalence of necrosis in patients with IMNM as the ACR/EULAR classification is unable to differentiate IMNM from PM. Necrosis, often with or without minimal inflammatory infiltrates, is characteristic of IMNM, reported in 93% of patients.31 For several years, muscle biopsy was considered as the “gold standard” for IIM diagnosis and served as a cornerstone for several classifications, particularly the ENMC classification.18 With the emergence of MSAs, diagnostic algorithms are evolving in favor of non-invasive methods, and it is suggested that biopsies must be reserved for seronegative IIM cases.32
Cardiac involvement in IIM varies widely in studies, ranging from 6 to 75%, depending on the inclusion or not of asymptomatic forms in the selection criteria.33 Myocarditis, reported in around 30% of autopsy series, is the most frequently described histological abnormality.34 As it is often subclinical, it was less common in our cohort. Electrographic abnormalities are the most common subclinical signs, accounting for 30 to 80% of cardiac manifestations,35 with the most common being bundle branch blocks, in line with our results.36
ILD was reported in 45% of our patients. Its frequency in the literature varies between 20 and 86%, depending on the diagnostic methods and the sample sizes of the different subgroups of IIM included. Consistent with other studies, ILD most presented with dyspnea and dry cough. However, its presentation is highly variable, ranging from asymptomatic forms to acute respiratory failure.37 This latter presentation holds a diagnostic challenge as it can mimic infectious pneumonia. Therefore, it is advisable to investigate for myopathy in cases of acute ILD without obvious pathological context.38 Four distinct histological aspects have been described in lung biopsies: cellular and fibrosing NSIP, OP, UIP and diffuse alveolar damage (DAD) and seems to be correlated with radiological patterns.39 NSIP is the most commonly reported pattern, found in 59% of our patients with ILD is reported in 61% to 81% of cases.40 Thus, heterogeneity in ILD behavior is well established, and several clinical presentations associated with specific histological and evolving patterns have been discerned.41 Given the invasive nature of lung biopsy, not recommended in this context, high-resolution thoracic computed tomography is the modality of choice for diagnosing and classifying ILD into patterns as defined by the American Thoracic Society/European Respiratory Society.42 Given the correlation between histological and radiological aspects, the most common CT abnormalities are ground-glass opacities and linear reticulations, corresponding to the NSIP pattern, in line with our study.39
Skin involvement in IIM is highly diverse. Cutaneous manifestations, crucial for DM diagnosis, have traditionally been categorized based on their diagnostic relevance and frequency into seven categories.43,44 Nailfold capillaroscopy, commonly used in SS patients, has been widely evaluated in IIM. Among CTDs, IIM exhibit the highest incidence of scleroderma-like pattern, second only to SS.45 Poikiloderma and periorbital/facial edema are considered compatible with DM.43 However, considered typical of DM, have been reported in our ASS patients, explaining their classification into the DM subgroup according to ACR/EULAR criteria. Palmar hyperkeratosis characteristic of ASS, is observed in 19 to 57% of cases.30 Similarly, although reported in two of our patients classified solely as DM, it was much more common in the ASS subgroup. Recently, hyperkeratotic lesions referred to as “hiker’s feet,” have been reported in both DM and ASS.30
Joint involvement noted in 48% of our patients are reported in 18% to 53% of IIM patients.46 These disparities can be explained by different definitions (arthralgia vs. arthritis) and varying inclusion criteria, impacting the composition of included subgroups, especially that of ASS. Although joint involvement is observed across other subgroups, it is more common in ASS.46 Consistent with the literature, polyarticular involvement was predominant (68%). Joint involvement is mild and rarely erosive.47
While IIF on HEp-2 cells to detect ANA is a standard screening method for CTD, its applicability in the context of myositis is debatable. It is not sufficiently sensitive to detect all ASA, as indicated by recommendations from the International Union of Immunological Societies and the European Autoimmunity Standardization Initiative.48 This lack of sensitivity is reflected in negative ANA results in 24% of our patients. Furthermore, there is no consensus regarding whether to consider antibodies directed against cytoplasmic components of HEp-2 cells as positive ANA, despite cytoplasmic staining enhancing ASA sensitivity. Moreover, even in cases of positive ANA, the commonly used algorithm for additional tests typically includes only anti-Jo-1 antibodies. Therefore, a multi-specific immunodetection assay is the recommended immunological test when suspecting myositis. Nevertheless, IFI remains valuable for several reasons. First, it remains the reference method in the frequent situation of overlap with CTD, especially SS. Additionally, since several MSAs exhibit distinctive nuclear or cytoplasmic IFI patterns, it can add specificity to a positive result obtained through immunodetection by confirming a pattern consistent with the MSA in question.49 Although these IFI aspects are not specific for most MSAs, this added value has been demonstrated for anti-SRP antibodies.50 Lastly, some myositis patients have positive ANA in IFI without any of the currently known MSA, suggesting that several antibodies have not yet been identified.49
More than 20 autoantibodies have been identified in patients with myositis, conventionally classified into Myositis Specific Antibodies (MSA) and Myositis Associated Antibodies (MAA), the latter referring to autoantibodies that can also be found in other CTDs. Interestingly, detecting multiple MSAs in the same patient is extremely rare, making them ideal biomarkers to identify homogeneous subsets of myositis.51
Most MSAs have been identified using the immunoprecipitation technique, the gold standard for ASM detection. Currently, new mono- and especially multispecific tests, readily available in the form of commercial kits, are on track to become the standard method for MSA detection. However, studies comparing these tests with immunoprecipitation or with each other have shown varied results.49 Vulstke et al., in comparing the performance of three multispecific tests (Euroimmun, Trinity, and Alphadia), demonstrated considerable variability in terms of specificity and sensitivity.52 Therefore, the standardization of these tests, through large prospective studies, is essential.53
MSAs are detected in 30 to 58% of IIM cases.54 They were found in 52% of our patients. This result seems underestimated, as only 11 patients were tested for all currently known MSAs. One consequence of the lack of standardization is the emergence of a significant number of multi-positivity cases, reported in 16.5% of patients tested by immunoblot compared to only 0.2% by immunoprecipitation. In such cases, a critical interpretation of immunoblot results, along with comparison to Hep-2 IFI data and clinical presentation, is recommended.55 This was the case for 8% of our patients.
Cancer was diagnosed in 17% of our patients, most commonly during DM, in line with existing literature. The frequency of cancer in IIM varies widely between 3 and 63%.56,57 DM has the highest increased risk, with a standardized incidence ratio ranging from 2.2 to 7.7 according to studies. Cancer can precede, accompany, or follow the diagnosis of IIM, but most cases occur concurrently or within a year of IIM diagnosis.58 Although it gradually decreases, the standardized incidence ratio remains elevated for several years.59 In an Australian study, it did not return to that of the general population even after 16 years of follow-up.60 Due to this temporal proximity, the term “Cancer Associated Myositis (CAM)” has been coined to define cancers diagnosed within three years of IIM diagnosis.58 This was the case for three-quarters of our patients. Consistent with the literature,61 adenocarcinoma was the most frequent histological type in our study. Lung, digestive, ovarian, breast, cervical, bladder, prostate cancers, as well as Hodgkin lymphomas, are among the most frequently associated cancers58 with varied frequencies, generally reflecting those observed in the matched general populations based on demographic characteristics.59
IIM are a heterogeneous group of inflammatory disorders affecting muscle with systemic involvement. Many classifications are proposed but none is exhaustive. Actually, the most used classifications are based on serological testing which contribute which contributes to the identification of the type IIM, its clinical phenotype and the association with other pathologies in particular neoplasia.
This work highlighted the polymorphism of IIM and revealed the inadequacy of current classification criteria, aligning with several other studies and joining experts in calling for standardization of immunodiagnostic methods and a clinico-serological approach. Such standardization would allow for harmonizing studies and better identifying IIM.
Our study has multiple limitations. It is a retrospective study. Data, such as muscle testing across all muscle groups, electromyographic data, histological data, and particularly a detailed description of the latter, were not consistently recorded. The monocentric and retrospective nature affected the number of seropositive IIM patients, as Dot-Myositis was not always performed. Additionally, the panel of MSAs tested was not complete in only eight patients, due to availability issue.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)