Keywords
Osteoarthritis (OA), Obesity, Mechanobiology, Joint Overload, Gut–Joint Axis, Microbiome Dysbiosis, Epigenetic Reprogramming, MicroRNAs (miRNAs), Inflammation, Early-Onset Osteoarthritis
Osteoarthritis (OA), Obesity, Mechanobiology, Joint Overload, Gut–Joint Axis, Microbiome Dysbiosis, Epigenetic Reprogramming, MicroRNAs (miRNAs), Inflammation, Early-Onset Osteoarthritis
Osteoarthritis, a destructive joint disease with cartilage degeneration and restructuring of subchondral bones is conceptually considered a synonymous disease in older adults.1,2 In recent years there is increased awareness of emergence of early-onset of OA often in the context of obesity and metabolic dysfunction.3 Obesity is one of the most significant modifiable risk factors for OA,4,5 therefore, epidemiological studies indicate that a moderate rise of body mass index (BMI) associates a large surge in risk of knee OA6 and doubles one’s lifetime risk.7 Importantly, obesity is a predisposing factor not only in weight-bearing joints (knees, hips, spine) but also in non-weight-bearing joints such as in the hands. This observation indicates that systemic aspects other than increased load are involved in OA in obesity. Obese adipose tissue releases pro-inflammatory cytokines and adipokines (e.g TNF-alpha, IL-6, leptin) that cause a systemic state of inflammation.8 These factors may prime joints for OA development and some researchers have begun to conceptualize a metabolic OA phenotype or obesogenic joint,9 whereby metabolic, inflammatory, and mechanical factors work together to promote OA development. It is plausible that the occurrence of early-onset OA in the obese patients is the result of this combination of excessive mechanical loading and systemic metabolic insults at younger age than typical idiopathic OA.
New developments have now widened the horizon on the obesogenic joint. Mechanobiology research findings indicate the severity by which abnormal forces exerted on cartilage and bone in the obese joint activates negative cell signaling cascades.10 Meanwhile, new studies of the gut-joint axis reveal that microbiome alterations associated with obesity may cause a systemic inflammatory effect and mechanically affect the joint through microbial metabolites. The impact of obesity and inflammation on OA has been investigated to reprogram chondrocyte and synoval cell gene expression by DNA methylation, histones modifications, and non-coding RNAs and may endure and continue to propagate the OA progression.11 These individual mechanisms are not independent of each other and in effect are a chain of interacting interfaces ( Figure 1). Inflammatory mediators have the potential to increase the mechanosensitivity of cartilage and affect epigenetic signatures whereas mechanical cartilage injury can liberate factors that can control local inflammation.12
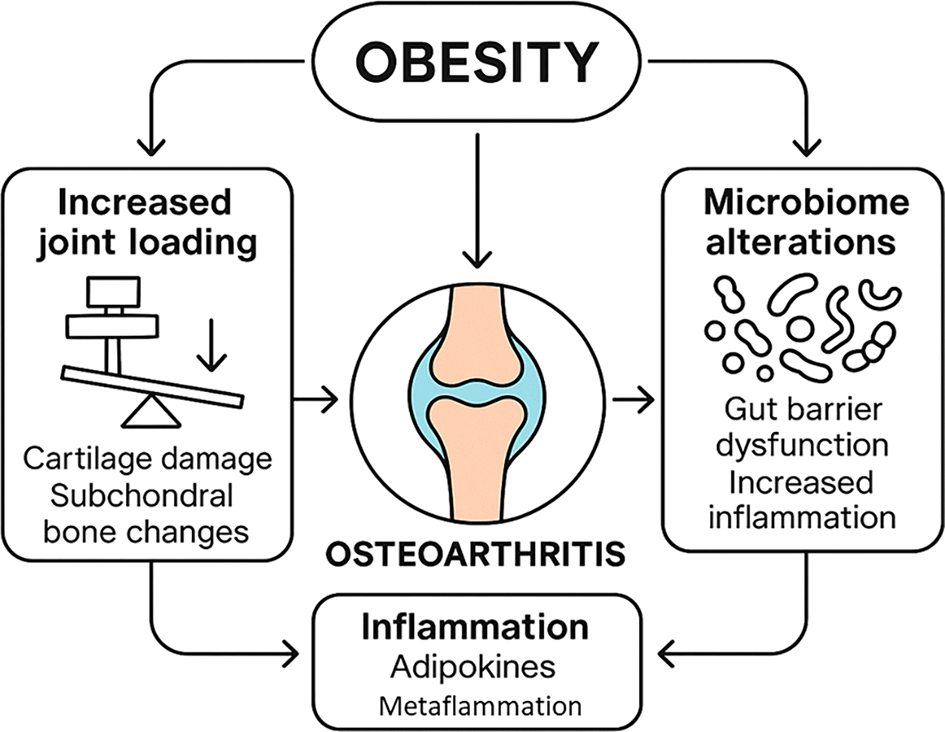
Obesity contributes to osteoarthritis via joint loading, microbiome alterations, and chronic inflammation, leading to cartilage damage, bone changes, and systemic dysfunction.
The aim of this review is to provide a comprehensive overview of the mechanobiological, microbial, and epigenetic factors that have been found to relate obesity with the early-onset OA. We reviewed the morphology of joint tissue at the cellular level and how it is influenced by biomechanical overload in obesity. We then discussed why gut and joint microbiome dysbiosis is likely to contribute to inflammation and cartilage breakdown and elaborated on epigenetic changes in OA and their potential obesity-related effects. An integrative perspective is provided to further combine these interfaces into a single pathogenic framework. Lastly, the review highlighted clinical implications, such as current and future potential mechanisms of diagnostic biomarkers and therapeutic agents targeting these mechanisms and offer future directions on research and management of obesity-related OA.
This review was conducted as a narrative synthesis of the literature on the mechanobiological, microbial, and epigenetic interfaces linking obesity with early-onset osteoarthritis (OA). While not a systematic review, transparent procedures were followed to enhance credibility and reproducibility.
The electronic searches were carried out in PubMed, Scopus and Web of science between January 2010 and June 2025 and further targeted searches within Google Scholar to include any grey or cross-disciplinary literature. Keywords such as “obesity”, “osteoarthritis”, “mechanobiology”, “gut microbiome”, “Epigenetic Reprogramming”, Inflammation” “Early-Onset Osteoarthritis” and gut-joint axis were used to search on the databases.
The initial search provided about 480 entries in the databases. Once the deduplication of the articles had occurred, 402 remained. The title and abstract screening excluded 237 studies as not relevant. Full-text review of 165 articles resulted in the inclusion of 51 studies for synthesis, representing the most relevant and current evidence base on mechanobiological, microbial, and epigenetic factors in obesity-related OA.
Data extraction and synthesis
The essential information from the research was acquired using a well-organized form: The extracted information include:
• Author(s)
• Year of publication
• Title of the study
• Country/region of study
• Study type/design (e.g., clinical trial, cohort, cross-sectional, case-control, animal experiment, in vitro)
• Population details:
• Mechanobiological aspects studied (e.g., joint loading, cartilage stress, adipokines, inflammatory mediators)
• Microbial aspects studied (e.g., gut microbiome changes, microbial metabolites, dysbiosis–inflammation link)
• Epigenetic aspects studied (e.g., DNA methylation, histone modification, miRNAs, noncoding RNAs)
• Biomarkers/outcome measures assessed (e.g., cytokines, matrix metalloproteinases, epigenetic markers)
• Key findings on OA onset or progression related to obesity
Excess body weight puts extra forces on joints with each step and each movement. Obesity is a factor in the pathogenesis of cartilage degeneration via biomechanical overloading.13 Clinical studies have correlated increased BMI with increase in cartilage volume loss over time on Magnetic Resonance Imaging (MRI) and earlier in the establishment of radiographic OA changes.14 In addition, obese individuals are usually characterized with altered gait kinetics and can also develop varus knee alignment thereby increasing the stress on medial compartments of knee. The menisci and subchondral bone are also subjected to more loads and result in the menisci tears and bone marrow lesions which add on the joint damage. Therefore, mechanical factors give the first kick in obesogenic joint leading to the destruction of cartilage and injuries of joint surface at a younger age ( Figure 2). Chondrocytes are very sensitive to the intensity and the frequency of mechanical loading at cellular level. In physiological conditions, mechanical stimuli contribute to cartilage homeostasis by signaling to induce matrix synthesis and to suppress catabolic enzyme expression.15
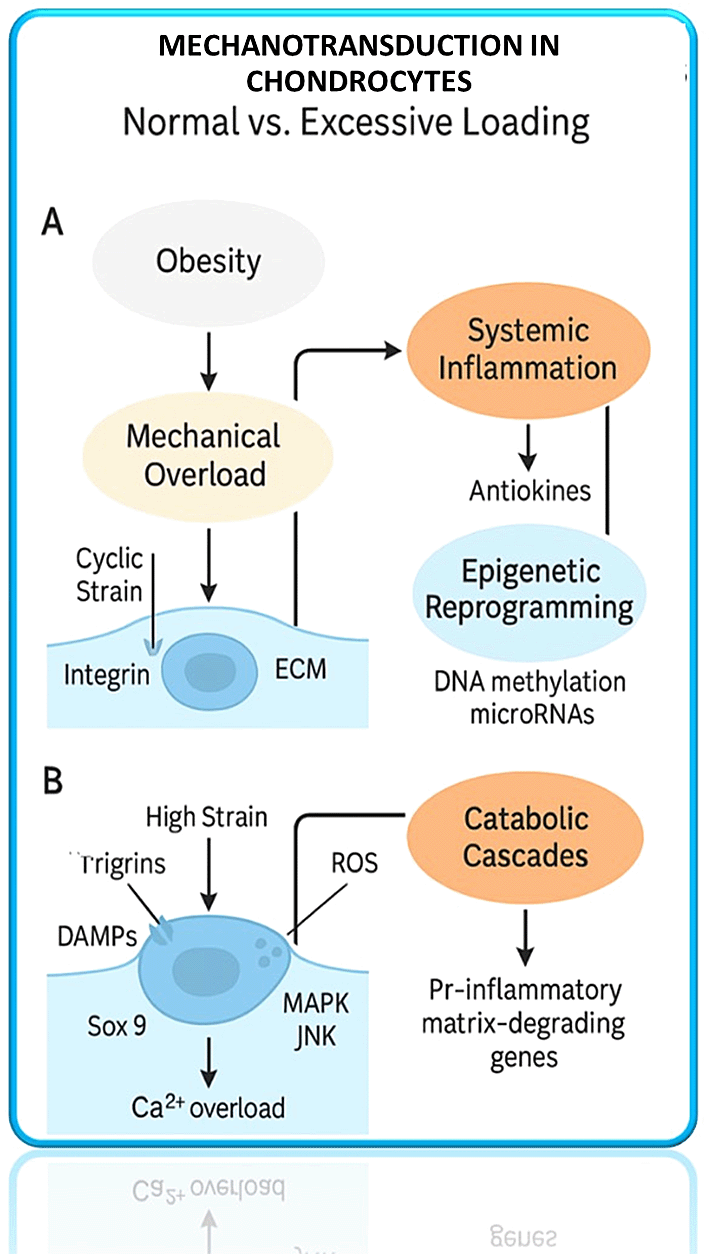
Obesity-driven mechanical overload and systemic inflammation trigger integrin signaling, ROS generation, and epigenetic reprogramming, leading to pro-inflammatory and matrix-degrading gene expression.
But when loading is excessive or abnormal (as in obesity or malalignment), this activates aberrant signaling in chondrocytes that causes them to become pro-degradative and inflammatory.16 Figure 2 in vitro and animal model experiments indicate that high-magnitude cyclic loading on cartilage increases the expression of matrix-degrading enzymes such as matrix metalloproteinases (MMPs) and aggrecanases, and decreases the expression of anabolic genes (e.g. collagen II and aggrecan).17 One of these mechanistic pathways is the mechanosensitive induction of gremlin-1. This is a BMP (bone morphogenetic protein) antagonist in chondrocytes exposed to injurious load. Gremlin-1 has been demonstrated to activate NF-kB pathway resulting in the upregulation of MMPs and A Disintegrin and Metalloproteinase with Thrombospondin Motifs (ADAMTS) enzymes which degrade cartilage matrix.18 Inhibition of gremlin-1 in mice (through neutralizing antibody or cartilage-specific gene deletion) reduces OA development under mechanical stress, which demonstrates its role as a mechano-inducible catabolic factor.19 Oxidative stress in chondrocytes can also be caused by excess load - Reactive Oxygen Species (ROS) production increases with high strain, which can lead to activation of Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells (NF-kB) and Mitogen-Activated Protein Kinase (MAPK) pathways that converge on pro-inflammatory and catabolic gene expression.18 In effect, the obese joint engages in maladaptive mechanotransduction: integrin, stretch-activated ion channel (e.g. PIEZO, TRPV4), and cytoskeletal signals activate transcription factors such as NF-kB and AP-1 to induce cartilage degradation. At the same time, protective mechanisms, which usually react to mechanical stimulation (the activation of anabolic transcription factors or anti-inflammatory mediators), are overwhelmed or dysregulated. In another example, excessive mechanical loading may inhibit TGF-/BMP signaling in cartilage by increasing BMP antagonists such as gremlin-1, as mentioned above, and by triggering inflammatory cytokines that disrupt TGF- anabolic actions.20
Mechanical strain as a result of obesity does not only impact the cartilage but also the subchondral bone and synovium. Chronic elevated joint loads cause subchondral bone remodeling, which starts with bone marrow edema and microfractures and is followed by sclerosis (thickening) of the subchondral plate. A stiffer subchondral bone, in turn, distributes stress less uniformly, leading to more focal cartilage damage. This produces a vicious cycle of degeneration. Meanwhile, mechanical damage to cartilage (even microscale injury) may trigger the release of danger-associated molecules (DAMPS) into the joint cavity, which induces synovial inflammation.21 In fact, obese patients with OA may have some degree of synovitis (detectable by MRI or arthroscopy), synovial macrophage infiltration and increased levels of synovial fluid inflammatory mediators. Joint overloading may, therefore, modify the synovial milieu to become a source of pro-inflammatory cytokines (such as IL-1β and TNF-α)22 that in turn act on cartilage and bone.23,24 This is a combination of direct cartilage mechanotrauma and secondary inflammation caused by tissue injury and is sometimes called mechanoinflammation. In obesity, mechanoinflammation can be increased due to systemic inflammatory priming, i.e. that minor mechanical insults can trigger a more pronounced synovial response than in a non-obese joint.
Although mechanical factors trigger the process of joint degeneration, the effects of obesity on OA go beyond the joint itself. One of the recent revelations in the knowledge of systemic factors in OA has been the presence of a gut-joint axis, whereby changes in the gut microbiome that occur with obesity lead to joint pathology25 ( Table 1). Obesity is usually associated with gut microbiota dysbiosis, which is the loss of microbial diversity and changes in the proportion of specific bacterial taxa. Typically, dysbiosis associated with obesity entails an elevated Firmicutes/Bacteroidetes ratio ( Figure 3) and loss of beneficial commensals (including Bifidobacteria) and an expansion of microbes that drive inflammation25 ( Table 1). Such dysbiotic microbiota can weaken the intestinal epithelial barrier (leaky gut) and cause translocation of bacterial products, such as lipopolysaccharide (LPS), into the circulation. The resultant metabolic endotoxemia leads to systemic inflammation due to upregulation of innate immune receptors (e.g. Toll-like receptor 4 on macrophages), which has been implicated in insulin resistance and possibly in joints as well.26,27
| Microbial taxa/metabolite | Direction in obesity | Joint/pathogenic effect | Supporting evidence | Translational angle | Reference |
|---|---|---|---|---|---|
| Firmicutes/Bacteroidetes ↑ | Dysbiosis shift | Promotes systemic inflammation | Animal models, human fecal studies | Prebiotic/probiotic correction | 25,29,30 |
| Bifidobacteria ↓ | Loss of commensals | Reduced SCFA, leaky gut | Oligofructose studies in obese mice | Dietary fiber, probiotics | 25 |
| LPS (endotoxemia) ↑ | Circulating microbial product | Primes synovial macrophages, metaflammation | Human plasma & synovial fluid studies | Gut barrier restoration, anti-inflammatories | 26,27,35 |
| Secondary bile acids ↑ | Dysregulated bile acid metabolism | FXR–GLP-1 axis suppression, cartilage catabolism | Bile acid study | FXR antagonists, GLP-1 analogs | 33,34 |
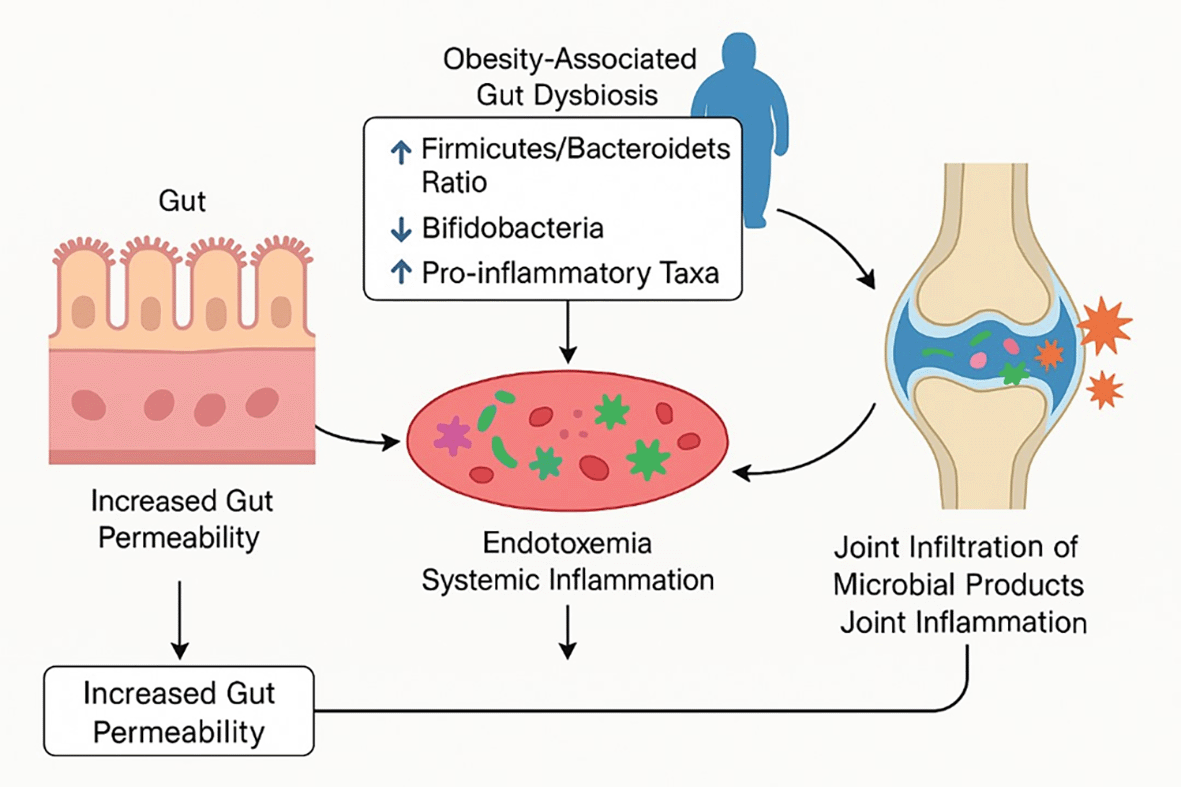
Obesity-related gut dysbiosis increases intestinal permeability and endotoxemia, driving systemic and joint inflammation that accelerates cartilage degeneration.
The gut- joint axis has been supported by animal studies. In an animal model of diet-induced obesity, dysbiosis was linked causally with OA progression.28 The mice with obesity had an increased level of pro-inflammatory intestinal bacteria and a decrease in Bifidobacteria, and a higher level of inflammatory products in the blood and faster rate of knee cartilage destruction25 ( Table 1). When these obese mice were treated with a prebiotic (oligofructose) to reconstitute a healthier gut microbiome, impressive outcomes were achieved. Bifidobacterium levels (most notably B. pseudolongum) increased, gut barrier integrity was enhanced, systemic and local (synovial) inflammation was reduced and the development of OA was inhibited25 ( Table 1). Oligofructose-fed obese mice exhibited fewer macrophages in the synovium and decreased levels of inflammatory factors in the knee, which was translated to fewer cartilage erosions than those in untreated obese controls. This indicates that the health of the joints can be directly affected by the modulation of the gut microbiota, at least in obesity. ( Figure 3) Similar trends have been found in other studies that have found correlations between some of the genera of the microbiome and the extent of cartilage damage.29 A study that used fecal transplantation in mice showed that an obesity-associated microbiome could exacerbate OA pathology whereas a lean microbiome was able to partially protect cartilage against damage despite controlling body weight differences25 ( Table 1). Particularly, it has been identified that certain microbes may be involved in this gut-joint interplay. In another study, nine microbial genera were found to be significantly related to cartilage histopathology regardless of diet and obesity, including Rikenella (positively associated with OA severity) and Blautia and Dorea (negatively associated, perhaps reflecting protective commensals).29,30 These associations are still being disentangled but they highlight the idea that microbial metabolites and antigens can have an impact on joint tissues.
A common way in which gut microbes are connected to joint degeneration is through circulating metabolites.31 Short-chain fatty acids (SCFAs) such as butyrate, which are produced by fiber-fermenting commensals, are mostly anti-inflammatory and keep the gut barrier healthy ( Table 1). Dysbiosis is a common factor that leads to a decrease in SCFA production, which causes inflammation. On the other hand, some metabolites produced by microbes can directly damage joints.15 Some of which include branched-chain amino acid metabolites or trimethylamine N-oxide (TMAO) produced by some bacteria in the guts of obese hosts, which have been shown to contribute to a systemic inflammatory state.32 ( Figure 3) More recently, a role in the gut-joint axis has been found in bile acid metabolism.33 In obesity, there is a shift in the bacteria of the gut that may alter the bile acids that stimulate the receptors of the host. A 2025 study found that elevated levels of some secondary bile acids (such as ursodeoxycholic acid derivatives) produced by gut microbes were able to stimulate intestinal farnesoid X receptor (FXR) signalling, which inhibited the secretion of a gut hormone (GLP-1) with chondroprotective properties.33 Genetic or pharmacologic inhibition of this FXR pathway increased GLP-1 secretion, which led to a reduction in OA development in obese mice. This observation suggests an advanced gut-microbe-metabolite-joint signaling: the intestinal FXR-GLP-1 axis. Increased GLP-1 (either via gut signaling or via therapeutic GLP-1 analogs) was demonstrated to have anabolic effects on chondrocytes and to decrease inflammation in the joint. Interestingly, GLP-1 receptors are expressed in articular cartilage and synovium and a GLP-1 agonist (liraglutide) in mice has been shown to protect cartilage and even with diabetic patients, it has an inverse correlation with OA progression.34 These observations demonstrate the close relationship between the microbiome of the gut and its metabolites and joint physiology
There is also emerging evidence that microbes (or their components) can travel between the gut (or other mucosal sites) to the joint. Even though joints were traditionally regarded as sterile sites, DNA of intestinal bacteria has been found in the synovial fluid or the synovial tissue of patients with OA.35 As an example, some osteoarthritic joint samples have been found to contain DNA of gut commensals, such as Prevotella or Bacteroides species, but not healthy controls.35 This may be due to bacterial translocation across a permeable intestinal lining into the bloodstream and implantation into the joint (particularly when the joint is inflamed and draws immune cells that may carry microbial DNA). Although these bacteria are not causing overt infection, their presence may be a persistent stimulus to the immune system within the joint causing ongoing synovial inflammation. Some researchers have suggested a distinct synovial microbiome in OA, where sequencing studies have identified low-abundance bacterial signatures in OA synovium distinct to those in RA or healthy joints.36 The clinical relevance of an intra-articular microbiome remains undetermined, but brings up interesting questions of direct microbe-joint interactions. It also offers a possible explanation of how obesity-related OA is systemic where events in the gut can affect a distant organ (the joint) through immune and molecular mediators.
Obesity may thus generate a pro-arthritic microbial signature, that is, a loss of beneficial species and an enrichment of pro-inflammatory species, that promotes OA through systemic inflammation through direct joint colonization or signaling by metabolites. This therapeutically provides an opportunity to target microbiome in OA.
Epigenetics is the study of heritable changes in gene expression that are not caused by changes in the DNA sequence. In OA, there is an increasing amount of evidence that epigenetic changes in chondrocytes and synovial cells play a role in the development and progression of the disease.37 These epigenetic modifications can be regarded as a memory of the past events of the environmental exposure, e.g. mechanical stress or inflammation that are imprinted on the cells and bias their behavior. Obesity, which presents a chronic inflammatory environment and metabolic disturbances, is an environmental factor that can cause such epigenetic reprogramming of joint tissues.
Among the first epigenetic discoveries in OA was altered DNA methylation of cartilage. In normal cells, the transcription of genes is normally inhibited by methylation of cytosines in CpG sites (especially in the gene promoter regions.38 In OA cartilage, catabolic and inflammatory genes are more likely to be hypomethylated (and thus overexpressed) and anabolic or homeostatic genes more likely hypermethylated (silenced). As an example, the promoters of a number of matrix-degrading enzymes - ADAMTS4 (aggrecanase-1) and MMPs 2, 3, 9, 13 have been shown to be demethylated at certain CpG sites in OA cartilage when compared with normal cartilage39,37 ( Table 2). This demethylation is associated with their upregulation in OA which directly leads to matrix breakdown. Conversely, genes that are critical to cartilage structure (such as collagen II or aggrecan) or anti-catabolic signaling may obtain aberrant methylation which inhibits their expression in OA. Such changes indicate that even when the inciting factors (mechanical injury, cytokines) are no longer present, the chondrocytes in an OA joint may be in a pre-programmed state biased towards catabolism. Obesity may enhance these changes: pro-inflammatory cytokines (IL-1, TNF-a) and adipokines (such as leptin) are found in obesity and may change the activity of DNA methyltransferases and demethylases in chondrocytes. In fact, targeted demethylation of MMP promoters can be induced by exposing chondrocytes to inflammatory cytokines, as has been demonstrated in studies40,41 ( Table 3). Similarly, elevated levels of leptin (typical in obesity) can epigenetically precondition chondrocytes to be more responsive. Chondrocytes in obese OA patients have been shown to overexpress MMP-13 in response to leptin, possibly because of epigenetic upregulation of the leptin signaling pathway in these cells.42
| Epigenetic layer | Altered marker | Change in OA (± obesity) | Functional consequence | Evidence | Potential therapeutic target | Reference |
|---|---|---|---|---|---|---|
| DNA methylation | ADAMTS4, MMP promoters | Hypomethylation → overexpression | Matrix degradation | OA cartilage methylome | Methylation modulators | 37,39,40 |
| microRNAs | ↓ miR-140; ↑ miR-22, miR-103 | Obesity tilts balance to catabolism | Loss of protective checkpoints | OA cartilage miRNA profiling | miRNA mimics/antagomirs | 45–47 |
| Histone modifications | IL-1β promoter hyperacetylation | Sustained inflammatory gene expression | Pro-inflammatory signaling | Chondrocyte histone assays | HDAC inhibitors, SIRT1 activators | 12,49 |
| Epigenetic layer | Target(s) /marker(s) | Direction in OA (± obesity) | Functional implication | Evidence | Potential interventions | Reference |
|---|---|---|---|---|---|---|
| DNA methylation | ADAMTS4; MMP-2/3/9/13 promoters | Hypomethylation → overexpression; inflammatory/adipokine milieu in obesity can drive demethylation | Matrix catabolism → cartilage degradation | OA cartilage shows promoter demethylation of catabolic genes; cytokines induce targeted MMP demethylation. | Anti-inflammatory upstream strategies; (conceptually) restore methylation balance. | 37,39,40 |
| Adipokine-linked epigenetic priming | Leptin → MMP-13 | Obese-OA chondrocytes over-respond to leptin (epigenetic priming) | Heightened catabolic responsiveness under obesity | Leptin-induced MMP-13 overexpression noted in obese OA chondrocytes. | Target leptin axis; reduce adiposity and leptin tone; leptin–mechanical synergy recognized. | 42 |
| microRNAs (protective loss) | miR-140 (targets ADAMTS5, IGFBP5) | ↓ in OA; obesity skews hostile miRNA milieu | Loss of catabolic “brakes” → early OA susceptibility | miR-140 abundant in healthy cartilage; ↓ in OA; knockout → early OA. | Concept of miR-140 mimic/local delivery discussed as future avenue. | 45 |
| BMI-associated miRNA shifts | ↑ miR-22, miR-103; ↓ miR-25, miR-29a, miR-337 | Obesity-correlated signature tilts toward inflammation/catabolism | Links adiposity to chondrocyte regulatory state | BMI-linked miRNA panel in knee cartilage (Iliopoulos et al.). | Anti-miR/mimic strategies (conceptual) as precision approaches. | 46,47 |
| Histone modifications | IL-1β promoter hyper-acetylation; HDAC/HAT imbalance; β-hydroxybutyrylation | ↑ access to pro-inflammatory genes; metabolic state (ketosis) rewires histone marks | Sustained inflammatory transcription; matrix loss | Histone acetylation changes in OA; IL-1β promoter hyperacetylation; metabolic β-hydroxybutyrylation link. | 12,49 |
The other important layer of epigenetic control is the microRNAs (miRNAs). These short non-coding RNAs (22 nucleotides) regulate gene expression in a post-transcriptional way by binding to target mRNAs, which causes their degradation or translational inhibition. Many miRNAs have been reported to be involved in cartilage homeostasis and OA pathophysiology43,44 ( Table 2). Interestingly, a number of miRNAs seem to have protective functions by keeping catabolic factors at bay and these are known to be downregulated in OA. A notable example is miR-140 which is one of the most abundant miRNAs in healthy cartilage and which targets ADAMTS5 and IGFBP5. miR-140 expression is severely reduced in OA cartilage and miR-140 knockout mice develop early-onset OA45 ( Table 2). On the other hand, some miRNAs that enhance inflammation or matrix destruction are increased in OA (e.g., miR-34a, miR-146a in some contexts, miR-22), and worsen the disease process. Notably, it has been shown that obesity is associated with certain miRNA alterations in OA. A study by Iliopoulos et al. profiled miRNA expression in knee cartilage and identified 16 miRNAs differentially expressed in OA vs. healthy cartilag46 Five of these miRNAs were associated with BMI of patients, with miR-22 and miR-103 being increased in obese patients with OA, and miR-25, miR-29a, and miR-337 being decreased with higher BMI47 ( Table 2). The miRNAs up-regulated in obesity (miR-22, miR-103) are believed to be harmful (e.g., inducing inflammation or cartilage catabolism), whereas those down-regulated (miR-25, miR-29a, miR-337) are chondroprotective. This BMI-related miRNA signature implies that obesity has the potential to shift the balance of cartilage miRNA expression to one that promotes OA. The downregulation of miR-140 in OA has also been confirmed in other studies, as well as the fact that miR-146a (which normally suppresses inflammatory responses) is negatively correlated with OA severity.48 These epigenetic and miRNA changes may give an answer as to why obesity may be a cause of accelerating OA at an earlier age, as the constant metabolic and inflammatory stress may lead to a faster pathological reprogramming of the joint cells.
Another epigenetic mechanism that is applicable to OA is histone modifications (acetylation, methylation, phosphorylation of histone tails). Histone acetylation is a process that tends to loosen chromatin and increase gene expression. It has been discovered that a cartilage has changed levels of histone acetylation on cytokine genes promoters and matrix genes promoters12 ( Table 2). As an example, the IL-1 beta promoter can be hyperacetylated in the OA chondrocytes, which encourages overproduction of IL-1. Obesity may affect these histone marks via metabolites. One interesting discovery is that β-hydroxybutyrate (an elevated ketone body during ketogenic diets or fasting) can be used as a substrate in a new histone modification (lysine β-hydroxybutyrylation) that has regulatory effects on gene expression.49 In an animal model, obese mice placed on a ketogenic diet had elevated histone β-hydroxybutyrylation levels in joint tissues and low OA progression.50 This suggests that there can be direct effects of systemic metabolic state (ketosis vs. high-fat diet) on the epigenetic landscape in the joint. Further, histone modifying enzymes such as histone deacetylases (HDACs) and histone acetyltransferases (HATs) have also been linked to OA. HDCs are usually hyperactive in OA, which leads to inappropriate gene silencing and a decreased expression of cartilage matrix components. Inflammatory signaling that is prevalent in obesity activates some HDACs. HDAC inhibitors are of interest as potential therapeutic agents to reverse deleterious histone changes in OA.
The OA has evidence of epigenetic acceleration of aging. Epigenetic clocks (models based on patterns of DNA methylation throughout the genome) indicate that OA cartilage appears older than the chronological age of the patients.51 This early epigenetic aging has also been found in blood cells of OA patients,51 pointing at a systemic factor. Obesity in itself has been shown to lead to epigenetic acceleration of age in certain tissues. Therefore, an obese 45-year-old with OA of the knees may have a cartilage with an epigenome similar to that of a significantly older person, which adds to the risk of early joint degeneration.
Obesity is thus able to rewire the epigenomic and non-coding RNA profile of joint tissues, creating a catabolic and inflammatory environment of chronicity. The epigenetic modifications create a memory of the effects of obesity, and may continue to drive OA development even when the causative factors (e.g., high weight or inflammation) are subsequently decreased. The awareness of these changes does not only enhance the knowledge of OA pathogenesis but also opens the door to new biomarkers and drug targets (such as disease-specific DNA methylation marks or miRNAs that could be measured in synovial fluid or blood, and enzymes that could be inhibited to reverse epigenetic lesions).
The described pathogenic pathways, namely, mechanobiological stress, microbial dysbiosis, and epigenetic reprogramming, are tightly interconnected in terms of the obesogenic joint. Instead of acting alone, these factors establish a positive feedback loop that leads to early-onset OA. Obesity effectively provides a metabolic-inflammatory platform against which joint injury is exacerbated.52 As an example, too much mechanical loading associated with obesity causes cartilage matrix damage and release of breakdown products (e.g., fibronectin fragments, hyaluronan fragments) which in turn activate pattern recognition receptors on synovial cells, causing inflammation. In an obese person, this local inflammation is augmented by systemic factors, a greater concentration of circulating IL-6, TNF-alpha, and adipokines, resulting in a stronger synovial inflammatory response than in a lean person.7 The synovial inflammation, in its turn, contributes to the aggravation of cartilage destruction due to cytokines (IL-1 beta, TNF) and proteases, and osteophyte formation due to the imbalances in growth factors such as VEGF and TGF-B. Therefore, biomechanical injury and inflammatory signaling in obesity-related OA are synergistic.53
Meanwhile inflammation caused by the gut microbiome may be regarded as another second hit that constantly activates the immune system. Circulating LPS in an obese gut sensitizes synovial macrophages, as well as macrophages in osteoarthritic cartilage lesions themselves (where subchondral bone marrow macrophages can infiltrate).7 This adds to the low-grade synovitis that is seen in a number of obese patients with early OA, as opposed to non-obese OA patients who may have less pronounced inflammatory signs. The stimulated macrophages secrete pro-inflammatory cytokines and chemokines to maintain a vicious cycle of inflammation and matrix destruction. In addition, certain microbial metabolites can have direct effects on chondrocyte behavior, such as some secondary bile acids, which can alter chondrocyte metabolism through the FXR/GLP-1 endocrine axis and thus connect gut dysbiosis with impaired cartilage anabolism. Thus, in obesity, the inflaming phenotype (age-like chronic inflammation) is exaggerated in joints, which leads to acceleration of degenerative processes.
More importantly, these inflammatory and mechanical insults are in part hardwired into joint cells by epigenetic changes. Repeated exposure to the obese environment induces chondrocytes and synovial fibroblasts to become pro-inflammatory and matrix-degrading, and to store this memory in the form of epigenetic modifications. When the DNA surrounding an MMP gene has been demethylated and the histones within that locus have been acetylated, the chondrocyte will have a tendency to continue overproducing that MMP even when the stimulus (e.g., IL-1) waxes and wanes.54 Equally, the absence of a protective miRNA such as miR-140 implies the absence of a checkpoint on some catabolic pathways. This epigenetic lock in does not only maintain the disease state but could be the reason that early interventions are important.
The interaction of the three interfaces may be exemplified by the part of adipokines like leptin. Leptin is secreted in large quantities by adipose tissue in obesity and travels to the joint through circulation and perhaps local synthesis in infrapatellar fat.55 Leptin plays two mechanistic functions: it may directly regulate chondrocyte metabolism (stimulating the expression of MMPs and inflammatory mediators) and it may also affect immune cells, promoting inflammation. The effects of leptin may be compounded by mechanical stress; it has been shown that leptin and mechanical load together have a more catabolic effect on cartilage than either alone.56 Furthermore, leptin signaling can intersect with epigenetic regulators e.g. leptin can activate STAT3 and NF-kB, and these in turn recruit histone acetyltransferases to promoters of target genes in chondrocytes, enhancing their expression.57 The unexpected observation that genetically obese mice that lack leptin (ob/ob mice) or leptin receptors (db/db mice) do not develop severe OA in spite of extreme weight underscores the importance of these coordinated messages. In those mice, the purely mechanical impact of weight does not lead to full-blown OA without the metabolic/inflammatory leptin signals. This means that the obesogenic joint is a product of multiple factors interacting rather than simply weight.58
These interfaces also share common downstream pathways in common tissue. A paradigmatic case is the NF-kappa B pathway, which is a key mediator of stress and inflammation responses. Mechanical strain (through ROS and integrin signaling) activates NF-kB in chondrocytess18 Inflammatory cytokines released by adipose tissue or synovium also activate NF-kB; and even some microbial molecules (such as LPS) signal through NF-kB in immune cells. NF-kB then regulates the transcription of numerous genes that are of concern to OA (MMPs, ADAMTS, IL-6, etc.). Similarly, both biomechanical stress and cytokines can activate the MAPK pathways (Extracellular Signal-Regulated Kinase (ERK), p38, c-Jun N-terminal Kinase (JNK), which promotes Activator Protein 1 (AP-1)-mediated expression of catabolic genes.59 Such shared pathways imply that mechanical and biological factors cannot be cleanly dissociated in their actions they both activate the same deleterious cascades in the joint.
The joint overload and systemic inflammation of the obesogenic joint triggers a feed-forward loop: local cartilage damage and synovitis cause epigenetic changes that perpetuate catabolism and sensitivity to mechanical load and inflammation. This causes a faster development of OA than would otherwise be the case, typically in multiple joints. Consequently, obese patients often have OA in more than one location (polyarticular OA). Epidemiological studies indicate that 84 percent of individuals with OA report at least two affected joints, in line with a systemic driver such as obesity.60
Diagnosis and biomarkers: The multifactorial nature of obesity-induced OA creates new opportunities of early diagnosis and risk stratification. OA is traditionally diagnosed based on clinical examination and radiographic alterations that are frequently only evident when cartilage loss is extensive. In an obesity setting, one of the objectives is to recognize those at risk of early-onset OA, prior to severe structural alterations, with biomarkers of the mechanobiological, microbial, or epigenetic disturbances. As an example, systemic inflammation measures such as high-sensitivity C-reactive protein (CRP) are slightly elevated in obese individuals and have been linked to greater risk of knee OA progression, but CRP is not joint-specific. Interesting results have been reported in recent studies concerning the occurrence of adipokines and cytokines in serum and synovial fluid: high concentrations of synovial leptin and IL-6 are strongly correlated with increased cartilage destruction and pain in patients with osteoarthritis.61 A patient with knee pain, with an adipokine profile (high leptin, high resistin, low adiponectin ratio) would likely have a metabolic-OA phenotype that is likely to deteriorate. Also, indicators of intestinal permeability like plasma LPS or intestinal fatty acid binding protein may reflect a leaky gut that is a source of joint inflammation.
On the epigenetic front, circulating microRNAs have become a source of interest as a minimally invasive biomarker. Circulating miRNAs (in blood plasma or in exosomes) are differentially expressed in OA patients relative to healthy controls, some of which are associated with obesity. As an example, the level of miR-140 and miR-146a in plasma has been negatively correlated with the severity of OA in the knee,62 possibly due to their downregulation in the affected joint. A low plasma miR-140 in an obese patient with knee pain may indicate the onset of cartilage destruction. Similarly, an epigenetic change in blood cells (as a surrogate measure of a systemic epigenetic shift) or in other readily accessible tissues could serve as a biomarker of a faster OA process. The use of multi-omics is being investigated: the combination of microbiome analysis (stool sequencing of dysbiosis patterns), metabolomics (looking at metabolite patterns such as elevated bile acids or reduced Short-Chain Fatty Acids (SCFAs), and inflammatory profiling to create a multi-omics risk score of obesity-related OA. Although these sophisticated diagnostics are not in clinical practice yet, they are the translational effort to detect at risk patients early. Such biomarkers may soon enable clinicians working with obese patients to determine which patients may be candidates of intensive preventive interventions (e.g. weight loss programs, joint offloading, etc.) before the onset of irreversible joint damage occurs.
Imaging techniques are also coming up to capture the characteristic of metabolic OA. MRI can show synovitis and edema associated with inflammation that will not be seen on X-ray. In obese OA patients, there is a tendency to display MRI evidence of diffuse synovial thickening and effusion even at moderate stages of OA, which can be monitored. New PET tracers that target activated macrophages or matrix turnover are in development and may be able to help visualize the inflammatory burden in an obese arthritic joint to guide therapy.63
Therapeutic strategies: Treatment of OA in obese patients should be multi-modal and should take care of the local joint problems as well as the systemic factors. The key to it all is weight management. Weight loss has undisputed advantages of alleviating symptoms and improving the rate of OA progression. A landmark randomized trial has shown that a 10 percent weight loss in overweight/obese adults with knee OA resulted in approximately a 50 percent decrease in pain and improvement of function over 18 months. More weight loss is also associated with even better outcomes - patients who lost >20% weight had improved pain relief and quality of life to an even greater extent. Losing weight relieves the joint mechanically (one pound lost equals approximately four pounds less on the knee per step, and tends to reduce systemic inflammation (decreases in CRP, IL-6, and leptin64–67 ( Table 4). In this way, weight loss positively affects all the parameters of the obesogenic joint. Another important pillar is exercise, especially low-impact strengthening exercise which can enhance joint support due to strengthening of periarticular muscles. This decreases effective load on cartilage and has an anti-inflammatory effect through myokines produced by the muscle. Diet-induced weight loss combined with exercise has been demonstrated to have a synergistic effect in knee OA. Interestingly, weight control is also preventive: in young obese individuals who do not have OA, a permanent weight loss can significantly reduce the risk of future OA development. Clinically, intensive lifestyle programs or bariatric surgery (in those who qualify) are interventions that have not only systemic health benefits but also reduce OA pain and may postpone the need of joint replacement. Indicatively, obese patients with a history of bariatric surgery prior to knee replacement experienced reduced pain and improved functionality after the surgery compared to those who had no pre-operative weight loss. This indicates that the joint environment is modified in a positive manner by pre-operative weight loss.68
| Interface | Mechanistic target | Example intervention | Stage of evidence | Clinical potential | References |
|---|---|---|---|---|---|
| Mechanobiology | Joint overload, gremlin-1 pathway | Weight loss, biomechanical offloading, anti-NFκB | RCTs, animal models | Strong clinical feasibility | 18,64,66 |
| Microbiome | Dysbiosis, SCFA deficiency, bile acid signaling | Prebiotics, probiotics, FXR modulators | Animal, early human trials | Emerging translational area | 25,69,70 |
| Epigenetics | Aberrant DNA methylation, miRNA shifts, histone imbalance | HDAC inhibitors, miR-140 mimics, SIRT1 activators | Preclinical | High innovation potential | 72,73,75 |
In addition to weight loss and physical therapy, there is a range of pharmacologic and supplement approaches under investigation to target specific pathways in obesity-related OA:
Treatment of inflammation and adipokines: There are few disease-modifying agents in traditional OA, but the inflammatory component of metabolic OA opens the prospect of anti-cytokine therapy (similar to rheumatoid arthritis). Trials of IL-1 inhibitors in OA have been mostly negative in general OA patients, but it is believed that a subgroup of patients with high levels of inflammation (many are obese) would respond. One promising candidate is IL-6 blockade (e.g., with tocilizumab) as IL-6 is frequently elevated in obese subjects and mediates both local and systemic effects; small studies in erosive hand OA (an inflammatory OA subtype) have shown some efficacy of IL-6 inhibition69 This may be applicable to knee OA in obese patients with apparent signs of inflammation. Inhibition of TNF-alpha, a master inflammatory cytokine released by adipose tissue, has not shown robust effects on pain or structure in OA to date, though specific high-inflammatory-state patients may benefit- this is yet to be elucidated in trials. Concerning adipokines, no drugs are yet approved to directly neutralize leptin or resistin. Studies in animals have also employed leptin receptor antagonists or binding proteins to effectively inhibit OA progression, and leptin therefore remains a potential future target. Regulation of adiponectin - an adipokine with both anti-inflammatory and pro-inflammatory effects depending on the situation is complicated. Some OA therapies under development seek to increase the beneficial low-molecular-weight forms of adiponectin. Alternatively, downstream effects of adipokines could be targeted; e.g. as leptin increases VEGF expression in joints (leading to aberrant angiogenesis and pain), anti-VEGF drugs (such as bevacizumab) have been shown to alleviate post-traumatic OA in animal models.70 These immunomodulatory approaches are not yet mainstream practice, but they indicate how OA management may become more individualized, with obesity-related OA managed more as a mild inflammatory arthritis.
Microbiome-targeted therapies: Since a gut-joint axis has been shown to exist, it is appealing to implement interventions that correct dysbiosis. Prebiotic fiber supplement (e.g., oligofructose or inulin) has already demonstrated efficacy in obese mouse OA models,25,71 and human trials may be on the way. Dietary fiber supplementation and perhaps probiotics may re-establish a healthier microbiome that generates more anti-inflammatory metabolites. Limited studies in humans have suggested that probiotics can lower the systemic inflammation and pain in knee OA. To illustrate, a probiotic strain (Lactobacillus casei Shirota) administered to patients with knee OA led to decreased levels of serum IL-1 and improvement in pain over placebo in one study.72 FMT fecal transplantation of healthy donors to patients with metabolic syndrome is another experimental treatment, although the primary outcome measure is metabolic; this treatment may also improve joint health by decreasing systemic inflammation. Care should be taken, because the consequences of such modification of gut flora are not fully comprehended yet. A more specific intervention could be postbiotics (administration of beneficial microbial metabolites: e.g., SCFA enemas or butyrate supplements) or specific microbial enzyme inhibitors (e.g., inhibition of bacteria that produce harmful metabolites such as TMAO). Moreover, the fascinating Farnesoid X Receptor - Glucagon-Like Peptide 1 (FXR-GLP-1 gut-joint axis implies that bile acid modulators (e.g., FXR antagonists or enzyme inhibitors that alter bile acid composition) could protect the joint. A bile acid modulator (glyco-UDCA, an FXR inhibitor) attenuated OA in mice, which is a new line of therapy.73
Epigenetic therapies: The new concept of targeting the epigenetic changes in OA is in its initial stages. HDAC inhibitors (HDACi) such as trichostatin A or vorinostat have the potential to normalize the gene expression pattern of chondrocytes.74 HDACi have been demonstrated to decrease the expression of pro-inflammatory and catabolic genes in chondrocytes in cell culture and in animal models and to prevent cartilage damage75 ( Table 4). Systemic HDAC inhibition may have numerous off-target effects, thus work is underway to develop selective HDAC isoform inhibitors or local delivery. An example is that an intra-articular injection of HDAC6-selective inhibitor in a rat OA model decreased cartilage degeneration and pain indicating that local epigenetic therapy is a potential option.76 An alternative strategy is to exploit the body epigenetic regulators: SIRT1, a NAD+-dependent deacetylase, has generally chondroprotective effects (it deacetylates and inactivates p53 and NF-KB in chondrocytes, promoting survival and matrix synthesis). In OA, SIRT1 activity is decreased, especially in obesity where the excess of nutrients may inhibit SIRT1. SIRT1 activators (such as resveratrol or pharmacological activators) have demonstrated cartilage-protective effects in models and may be repurposed as OA treatments.77 In a similar manner, DNA methylation modulators are under investigation - such as dietary supplements like S-adenosylmethionine (SAM-e) which may facilitate methylation reactions and have demonstrated some symptom improvement in OA trials, though their mechanism is uncertain. The most precise epigenetic treatment in development is miRNA-based treatment. Based on the identified positive and negative miRNAs in OA, scientists are testing the use of miRNA mimics or inhibitors in the joint. As an example, a miR-140 mimic encapsulated in a safe vector could be injected intra-articularly to supplement this lost miRNA in obesity.78 An inhibitor (antagomir) of a deleterious miRNA such as miR-34a could slow cartilage degeneration.79
Traditional symptom management with a twist: Because disease-modifying approaches are under investigation, symptom control is paramount, particularly in early-onset OA where patients can live with decades of pain. Nonsteroidal anti-inflammatory drugs (NSAIDs) are frequently used as analgesics and they do affect inflammation. It is possible that obese patients with OA respond to NSAIDs better than non-obese patients because a larger proportion of their pain is of inflammatory origin. Nevertheless, there are cardiovascular and renal risks associated with the long-term use of NSAIDs, which are worrying in obesity. Therefore, adjuncts such as dietary anti-inflammatories (e.g., omega-3 fatty acids, which can decrease IL-1 production and which have been shown to have a modest beneficial effect on OA pain) may be advised. Nutraceuticals like curcumin (a turmeric extract) have NF-KB inhibitory effects and there is some evidence of improved pain and function in small studies- these may be especially useful in metabolic OA, where they can be used to address both metabolism and inflammation.80 Temporary relief is achieved by analgesic injections (with corticosteroids or hyaluronic acid). It is also of interest whether formulations can be optimized in obese patients. In younger obese patients, joint replacement is a problem because of the increased surgical risk and the possibility that the prosthesis may be worn out because of high loads. Thus, a focus on joint preservation modalities, potentially including new regenerative medicine (such as stem cell or platelet-rich plasma injections that have demonstrated some cartilage reparative potential), is of particular importance to this population.
In practice, it is best to have an individualized plan. An example would be a middle-aged obese patient with early knee OA; this patient could be treated with a combination of a weight loss and exercise program and a course of prebiotic supplementation to address dysbiosis. If signs of inflammation are present, a trial of an anti-IL-1 or NSAID to break the inflammatory cycle. The aim is to delay the progression of the disease and to control its symptoms in order to postpone end-stage interventions.
The unification of the fields of mechanobiology, immunology, microbiology, and epigenetics in OA research marks a more comprehensive study and treatment of the disease. Future studies are expected to be aimed at defining OA phenotypes (or endotypes) such as an obesity-driven OA subtype with specific biomarkers (high adipokines, dysbiosis, epigenetic markers of metabolic stress) and targeting them accordingly. Large cohort studies that combine multi-omics data are being undertaken to understand how factors such as diet, microbiome composition and epigenetic aging markers can predict OA outcomes in obese or non-obese individuals. It is possible to imagine predictive algorithms that, based on a patient microbiome profile, plus blood cytokine levels and genetic/epigenetic data, predict their 10-year risk of needing joint replacement, thereby initiating early aggressive treatment in those at high risk.
At the therapeutic level, there is a need to translate some of the promising targets that have been identified in preclinical studies into clinical trials. This would mean trials of prebiotics/probiotics to prevent OA in obese patients, trials of safe epigenetic drugs or gene therapies (e.g., intra-articular delivery of specific miRNAs or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) based editing to remove harmful epigenetic tags), and trials of metabolic therapies (e.g., GLP-1 receptor agonists) on joint outcomes. Interestingly, drugs such as GLP-1 agonists employed in diabetes/obesity (e.g., semaglutide) may be repurposed - initial epidemiological data suggest that diabetic patients on GLP-1 agonists have a reduced rate of joint replacements. A randomized trial is needed to determine whether these agents can delay the progression of OA even in the non-diabetic overweight patients. Metformin, another drug used to treat diabetes, activates AMP-Activated Protein Kinase (AMPK) (an energy-sensing enzyme) and has been shown to reduce inflammation and MMP expression in chondrocytes; retrospective studies have shown that diabetics taking metformin have slower knee OA progression than those not taking metformin81 This too should be tested prospectively as a low-cost intervention to target metabolic factors in OA.
The other future trend is regenerative and cell-based therapy to the obesogenic environment. Mesenchymal stem cell (MSC) injections are under investigation to heal cartilage, but in an inflammatory, obese environment, their effectiveness may be crippled.82 Researchers are studying preconditioning or genetic modification of such cells to be resistant to inflammatory cytokines or even deliver anti-inflammatory cytokines. Tissue engineering approaches may include the use of scaffolds that locally secrete an IL-1Ra (IL-1 receptor antagonist) or an Histone Deacetylase (HDAC) inhibitor to counteract the hostile environment in an obese joint and promote regeneration.
The evidence regarding the systemic effects of obesity on joints supports the importance of early prevention and treatment of obesity as a prevention measure against OA. Multidisciplinary clinics should treat patients in collaboration with rheumatologists, endocrinologists, and nutritionists. This will treat OA as part of a complex of metabolic diseases. Policy-wise, the acknowledgment of OA as an obesity-related condition (much like diabetes or heart disease) may result in the inclusion of joint health indicators in obesity management schemes.
In the context of basic science, areas of future research include cartilage and synovial methylome and microRNAome mapping in large patient cohorts to identify key regulatory nodes that can be targeted with drugs. An advanced method is CRISPR-based epigenome editing (e.g., using dCas9 fused to epigenetic modifiers to specifically demethylate or methylate a gene of interest in chondrocytes), which could potentially be used to functionally validate the role of specific epigenetic changes in OA. Should this work, it would be an intra-articular CRISPR treatment that would re-silence an inappropriately activated catabolic gene.
The other interesting field is the contribution of the joint microbiome. Because the presence of certain bacteria is constant in osteoarthritic joints, it is necessary to find out whether they are causative or opportunistic. Future development of antibiotics or phage therapy directed against specific joint-colonizing bacteria to treat a subset of OA is possible. Probiotics could be added to the joint to modify the local immune response.
Lastly, since it is a complex disease, prevention is better than cure. In young people with obesity, weight management and a healthy diet containing fiber and omega-3s could not only prevent diabetes and cardiovascular disease but also keep the joints healthy by ensuring normal mechanobiology, an intact gut microbiome, and a youthful epigenome in the cartilage. It should be confirmed in future longitudinal studies that the weight loss or diet improvement of those who lose weight or improve their diet reduces the risk of OA. This will be a crucial evidence base to guide the interventions in public health.
In conclusion, the obesogenic joint concept summarizes the multi-factorial effect of obesity on joint tissues, which is a combination of excessive mechanical loading, chronic inflammation (systemic and local), microbial effects, and persistent epigenetic alteration. The early onset OA in obesity is the result of this nexus. It will take similarly multi-dimensional solutions, a combination of lifestyle changes and specific biomedical treatments. By re-examining the osteoarthritic joint through the lenses of mechanobiology, microbiology, and epigenetics, we can not only gain a better understanding of disease pathology but also a more optimistic view on the potential novel treatments that could revolutionize care of the millions of people at risk of disabling arthritis because of obesity. There is a necessity to translate these findings to the bedside in terms of effective prevention and treatment of OA in the growing obese population.
All authors read and approved the final version of the manuscript and agree to be accountable for all aspects of the work.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)