Keywords
Network pharmacology, Molecular docking, medicinal plants, Phytochemicals, Drug discovery, Computational Biology,
This article is included in the Plant Science gateway.
This article is included in the Cheminformatics gateway.
This article is included in the AI in Medicine and Healthcare collection.
Network pharmacology, Molecular docking, medicinal plants, Phytochemicals, Drug discovery, Computational Biology,
In Version 2, the manuscript was revised to improve clarity, organization, and overall presentation prior to resubmission for further peer review consideration. The abstract, quality assessment, and data synthesis sections were refined for improved scientific clarity and readability. Several Results sections were also revised and reorganized for clearer presentation of findings, including sections on distribution of database categories reported, computational multi-database integrators, literature-derived compound sources, therapeutic domains, pathway and enrichment analysis tools, docked phytochemicals, reported binding affinities, and key molecular pathways identified across included studies.
Minor edits were additionally made to improve language, terminology, formatting, and consistency throughout the manuscript. An affiliation correction was also made for author Swase Dominic Terkimbi, who is affiliated with affiliations 1 and 8. No changes were made to the underlying study data.
Herbal medicine constitutes one of the oldest therapeutic modalities and continues to represent an important component of primary health systems globally.1 Increasing global interest in phytomedicine is driven by an increase in demand for natural therapeutic agents, recognition of ethnopharmacological knowledge, and the search for more safer compounds. Despite its extensive cultural and clinical relevance, progress in herbal medicine research has historically been constrained by challenges.2 These includes the complexity of multi-component formulations, limited mechanistic understanding, and insufficient molecular validation.3
Network pharmacology has emerged as a systems-level strategy for elucidating compound–target–pathway interactions and capturing the multi-target mechanisms characteristic of botanical therapeutics. Network pharmacology enables the interrogation of synergistic effects, prediction of key therapeutic nodes, and functional enrichment analysis by modelling biological networks and disease pathways.4 This approach aligns with the principles of traditional medicine, moving beyond reductionist drug discovery paradigms and offering a framework for evaluating multi-component interventions.5
Furthermore, complementary to network-based inference, molecular docking provides atomic-level understanding into ligand–protein binding interactions. This enables validation, target prediction, and pharmacodynamic potential assessment. Docking simulations allow estimation of binding affinity, identification of key residues involved in ligand recognition and prioritization of lead phytochemicals for downstream experimentation.6 When applied in combination, network pharmacology and molecular docking constitute an integrative in silico pipeline capable of accelerating mechanistic elucidation, drug-binding affinity, and phytochemical prioritization prior to laboratory validation.7,8
In recent years, an expanding body of literature has applied these computational approaches to medicinal plants and traditional formulations, contributing to deeper mechanistic understanding and discovery of promising bioactive compounds.9 However, the field remains methodologically heterogeneous, with variation in data sources, analytical workflows, validation strategies, and reporting standards. Consolidation of current evidence is therefore essential to map research trends, evaluate methodological rigor, identify knowledge gaps, and guide translational advancement. This systematic review synthesizes studies that have jointly employed network pharmacology and molecular docking to investigate herbal medicines and phytochemicals. The review aims to: (i) characterize methodological practices and computational resources used across studies; (ii) identify key therapeutic areas and biological pathways explored; (iii) assess the extent of experimental verification supporting computational predictions. Through this synthesis, the review provides an evidence-based foundation to inform best practices and enhance the translation of herbal bioactive into clinically relevant therapeutic candidates.
A systematic review was conducted in accordance with PRISMA 2020 and the PRISMA extension for Scoping Reviews (PRISMA-ScR). The protocol specified the research question, eligibility criteria, information sources, search strategy, screening processes, data extraction fields, quality appraisal approach, and synthesis plan before data collection commenced.
Studies were included if they met specific methodological and time-based criteria. Eligible papers were original, peer-reviewed research articles published between January 1, 2010 to 1st August, 2025. Each study was required to use both network pharmacology and molecular docking as part of its research approach, and the interventions had to involve herbal medicines, traditional plant formulations, or isolated phytochemicals. Studies also needed to report measurable scientific outputs such as compound–target interactions, protein network results, pathway or enrichment analysis, or docking scores showing ligand–receptor binding. For consistency and accurate interpretation, only studies published in English were considered. Studies were excluded if they did not use both network pharmacology and molecular docking together. Review articles, systematic reviews, meta-analyses, commentaries, letters, conference abstracts, and unpublished work were not included. Research focusing only on synthetic drugs without any herbal component was excluded. In addition, non-English publications and grey literature, such as theses and institutional reports, were not considered in this review.
A comprehensive literature search was conducted on 1st August 2025 to identify eligible studies. Three major scientific databases were searched including PubMed, Scopus, and Web of Science. A total of 83 records were retrieved across the three databases (PubMed = 26, Scopus = 10, Web of Science = 47). All articles were retrieved as CSV file and imported into Rayyan for systematic screening, duplicate detection, and management. During the de-duplication process, 9 duplicate records were detected. 4 duplicates were resolved and 5 was removed, leaving 78 articles for title and abstract screening. The search strategy combined controlled vocabulary terms and free-text keywords related to network pharmacology, molecular docking, and herbal medicine. The PubMed search string was: (“Network Pharmacology” [Title/Abstract] OR “Systems Pharmacology” [Title/Abstract]) AND (“Molecular Docking” [Title/Abstract] OR “In Silico Docking” [Title/Abstract]) AND (“Herbal Medicine” [Title/Abstract] OR “Medicinal Plants” [Title/Abstract] OR “Phytochemicals” [Title/Abstract]) AND (“2010/01/01” [Date-Publication]: “2025/12/31” [Date-Publication]). Equivalent search terms were adapted for Scopus and Web of Science to ensure consistency while accommodating database-specific syntax.
The study selection process followed a structured and transparent approach consistent with PRISMA-ScR guidelines. After the initial search and de-duplication, 78 articles were screened for title and abstract. Two reviewers independently assessed each article against the defined eligibility criteria. Articles that met the inclusion criteria were selected for full-text screen. During the screening process, any disagreements between the two reviewers were addressed through discussion. When consensus could not be reached, a third reviewer was consulted to resolve the discrepancy. Full-text articles were then examined to confirm compliance with the predefined inclusion criteria. Decisions at each stage of the screening process, including reasons for exclusion, were documented systematically. Out of the screened records, 36 studies met all criteria and were included in the final synthesis as depicted by Figure 1.
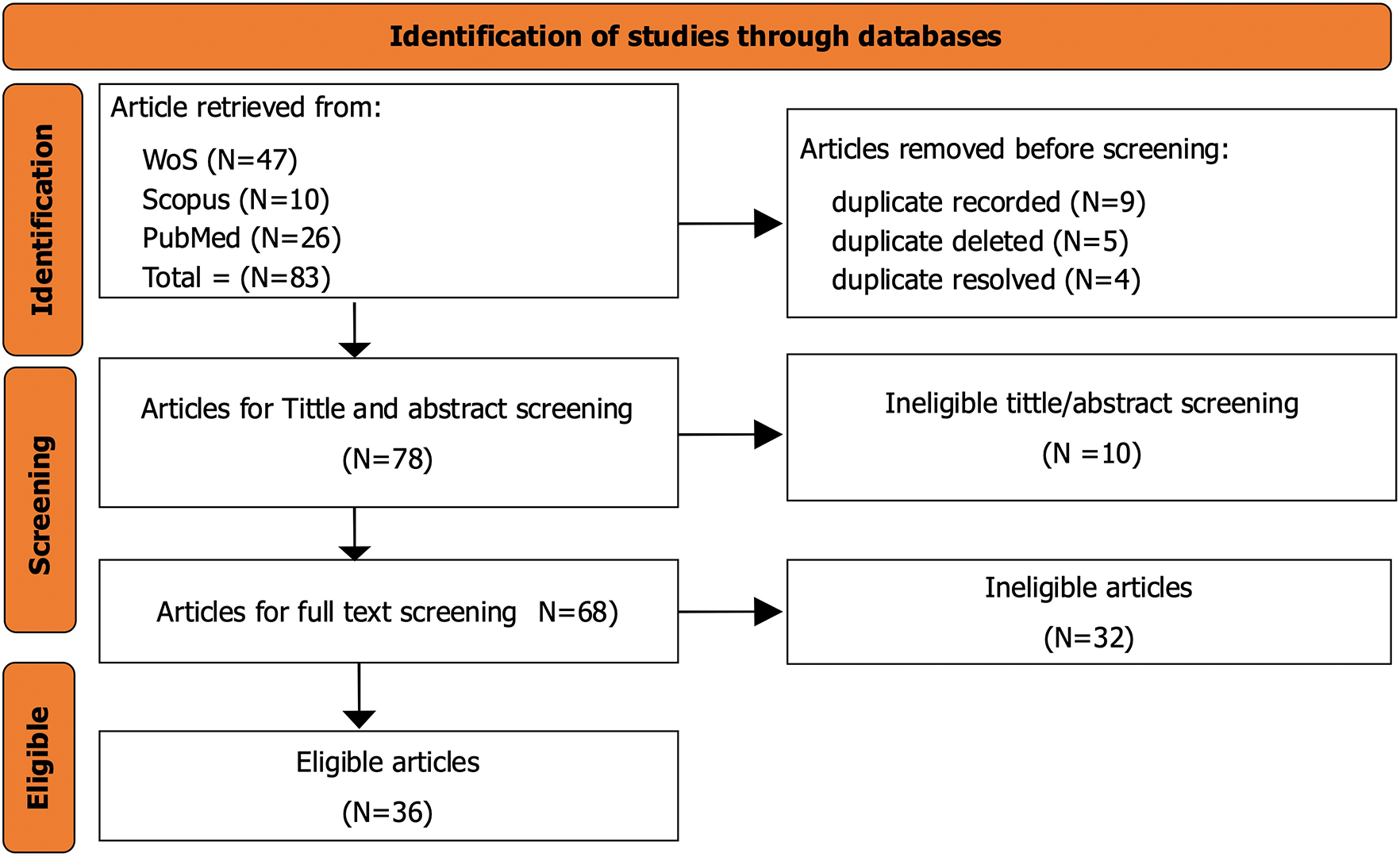
Data extraction was carried out using a structured and predefined data collection framework drafted in excel (version 2019) to ensure consistency and completeness. Key study characteristics and methodological details were systematically extracted from each included article. Extracted information included bibliographic data (author, year, journal, and country), herbal species or formulations investigated, plant part used, extraction or preparation method (where applicable), and the class of bioactive compounds assessed. Technical information relevant to methodologies and computational workflows was also collected. This included the databases used for compound identification and target prediction, software and algorithms applied in network construction, molecular docking tools and scoring functions, validation strategies, and major biological pathways or molecular targets identified. In addition, details related to experimental design, such as in vitro or in vivo validation, were captured when reported. The extraction process was conducted independently by two reviewers, and extracted data were cross-checked for accuracy. Any discrepancies were resolved through joint review and consensus discussion. Where information was incomplete or ambiguous, efforts were made to confirm details directly from study text and supplementary materials to ensure accurate interpretation.
Quality assessment focused on methodological transparency, rigor of computational approaches, and validity of outcomes. Studies were evaluated for reporting of databases, compound selection, target identification, network construction, docking protocols, scoring methods, and validation. Adherence to accepted standards in network pharmacology and molecular docking, including justification of tools, was assessed. Consistency with biological evidence and clarity of key outputs (compound–target interactions, pathways, docking scores) were also considered. Two reviewers independently appraised studies, with disagreements resolved by consensus or a third reviewer. Only methodologically sound studies were included.
A descriptive synthesis summarized study characteristics, herbal taxa, phytochemicals, disease indications, targets, and pathways. Frequencies and percentages were calculated for databases, prediction tools, network software, docking platforms, validation methods, and pathway resources, with totals normalized to 100% where applicable. Temporal trends (2010–2025), country distribution, and therapeutic areas were also summarized. Network maps of herb–compound–target–pathway relationships were generated where data permitted.
A total of 36 studies met the inclusion criteria and were incorporated into the final synthesis. The studies were published between 2013 and 2025, with a significant increase in publications from 2020 onward. Majority of the studies were conducted in Asian countries, particularly China (n = 20, 55.6%)4,10,11 and India (n = 9,12 25.0%), followed by Pakistan, Saudi Arabia, Egypt,13,14 Ethiopia,8 Indonesia, and Taiwan.15 Significant number of studies employed fully in silico approaches integrating network pharmacology and molecular docking (n = 22, 61.1%).12 The remaining studies combined computational pipelines with experimental laboratory validation, including in vitro assays (n = 8, 22.2%) and in vivo animal models (n = 6, 16.7%). Sample sizes for experimental arms varied, with most in vivo studies utilizing murine models, while in vitro analyses frequently employed human cancer cell lines and macrophage systems as summarized in Table 1. Plant materials analyzed varied widely and included whole plants, roots, rhizomes, barks, fruits,10,12 seeds, leaves, and formulated polyherbal preparations. Solvents used for extraction, where specified, included methanol,16 ethanol, chloroform, and n-hexane; however, solvent reporting was inconsistent across studies, with nearly half not specifying extraction media. Both single-herb studies (e.g., Centella asiatica, Piper longum, 10 Saussurea lappa) and polyherbal formulations (e.g., Huangqin Tang, Dashamoola, HSXFF, Krishnadi Churna, Qi-Enriching and Blood-Tonifying herbs) were reported. Biological validation where present included evaluation in disease-specific cell lines such as MCF-7 and A549, immune cells such as RAW264.7 macrophages and human peripheral blood cells, and rodent models of neurodegeneration, inflammation, ulcerative colitis, and metabolic dysfunction. Most purely computational studies validated predicted targets and pathways using publicly available protein structures, genomic databases, and docking or molecular dynamics outputs.
| ID | Author(s) | Year | Country | Sample Size | Study Type | Plant Part | Solvent | Medicinal Plant/Formula | Biological Source/Tissue |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 17 | 2024 | China | In vivo & in vitro (n not specified) | Experimental + NP + in vivo + in vitro | Not reported | Not reported | Centella asiatica/Asiaticoside | MPTP mouse model, BV2 cells |
| 2 | 18 | 2022 | India | 194 NCs screened | In silico NP & Docking | Not reported | Not reported | Clerodendrum sp. | Protein models |
| 3 | 19 | 2024 | India | 145 compounds | In silico NP & Docking | Fruit & root | Not reported | Piper longum | Fruit & root tissues |
| 4 | 10 | 2024 | China | Not specified | Experimental + NP | Fruit spikes | Ethanol | Piper longum L. | Mouse inflammation model |
| 5 | 12 | 2025 | India | Not specified | Experimental + Bioinformatics + NP | Fruits | Not specified | Caryota urens | MCF7 cells |
| 6 | 20 | 2025 | China | n=3 per group | Experimental + NP + Docking | Fruit | Not specified | Ficus hispida | HASMCs, mouse aorta |
| 7 | 16 | 2025 | China | n=6 per group | Experimental + NP + Proteomics + Docking | Roots | Methanol | Jiegeng Gancao Decoction | Colon tissue (UC mice) |
| 8 | 21 | 2025 | China | In silico | NP + Docking + Meta-analysis + DFT + MD | Roots & barks | Not specified | Dashamoola | Human intestinal GEO datasets |
| 9 | 22 | 2024 | China | In silico | NP + Docking + MD + DFT + ADME | Not specified | Not specified | Magnolia spp. | In-silico molecular models |
| 10 | 23 | 2022 | China | In silico | Bioinformatics + Docking | Rhizome | Not specified | Picrorhizae Rhizoma | Human genes |
| 11 | 24 | 2024 | China | In vitro | NP + Docking + Experiments | Roots/stems/rhizomes | Not specified | Huangqin Tang | MCF-7 & MDA-MB-231 cells |
| 12 | 25 | 2024 | Pakistan | In silico | NP + Docking + MD | Whole plant | Not specified | Cassia spp. | Target proteins |
| 13 | 26 | 2024 | India | In silico | NP + Docking + MD + MM-GBSA | Whole plant | Not specified | Drymaria cordata | Protein structures |
| 14 | 27 | 2024 | India | 40 phytochemicals | NP + Docking + MDS + MMGBSA | Root & Stem | Hexane/Methanol | Potentilla nepalensis | Cancer proteins |
| 15 | 28 | 2024 | China | A549 cells | Experimental + in silico | Root | Chloroform + Ethanol | Saussurea lappa | A549 lung cells |
| 16 | 29 | 2023 | Egypt | 32 plants, 2,154 compounds | NP + Docking + MD + in vitro | Whole plants | 70% Ethanol | Multi-herb panel | Human WBCs |
| 17 | 30 | 2023 | Saudi Arabia | Not specified | NP + Docking + MD | Not specified | Not stated | Dodonaea angustifolia | Not reported |
| 18 | 31 | 2023 | China | In silico | NP + Docking | Whole plant | Not specified | Solanum nigrum | Breast cancer genes |
| 19 | 11 | 2023 | China | In silico + in vitro | NP + Docking + CETSA | Whole plant | Not specified | Solanum nigrum L. | CT26 cells |
| 20 | 32 | 2025 | India | In silico + in vitro | NP + Docking + MD | Whole plant | Not stated | Alchornea laxiflora | Not stated |
| 21 | 33 | 2024 | China | In silico + in vitro | NP + Docking | Whole plant | Not specified | Polygonum cuspidatum | HEK-293T cells |
| 22 | 14 | 2022 | Saudi Arabia | In silico | Computational | Stem | Methanol | Argyreia capitiformis | Stem extracts |
| 23 | 34 | 2022 | China | In silico + experimental | NP + Docking | Seeds | Not specified | Rheum tanguticum | Seed tissue |
| 24 | 13 | 2021 | Saudi Arabia | In silico + in vivo | Experimental + NP | Leaves | Methanol | Cnesmone javanica | Leaf extracts |
| 25 | 35 | 2021 | Egypt | 100 compounds | In silico + in vitro | Propolis | Ethanol | Egyptian propolis | Propolis extract |
| 26 | 36 | 2022 | China | In silico | NP study | Whole plant | Not specified | HSXFF Formula | — |
| 27 | 15 | 2013 | China | In silico | NP modeling | Roots/rhizomes/tubers | Not specified | Qi-Enriching & Blood-Tonifying herbs | Molecular models |
| 28 | 37 | 2024 | India | In silico + in vivo | NP + Docking | Not reported | Not reported | Krishnadi Churna | Not stated |
| 29 | 38 | 2016 | China | In silico | Computational | Whole plant | Not specified | SH Formula | Not stated |
| 30 | 8 | 2022 | Ethiopia | 10 aloe species | In silico profiling | Leaves | Not stated | Aloe species | Leaves |
| 31 | 39 | 2022 | Indonesia | In silico | Virtual screening | Not specified | Not stated | Moringa & Psidium | Not specified |
| 32 | 7 | 2016 | Taiwan/China | Not specified | Virtual Screening | Not stated | Not stated | Multiple TCM herbs | Not reported |
| 33 | 40 | 2020 | Pakistan | In silico + in vitro | Phytochemical + Docking | Root | Ethyl acetate | Ziziphus oxyphylla | Not specified |
| 34 | 41 | 2021 | China | Not specified | NP + transcriptomics + in vitro | XYPI compound | Not reported | XYPI | RAW264.7 macrophages |
| 35 | 42 | 2025 | China | Not specified | In silico | Not stated | Not stated | BK002 formula | Patient genomic data |
| 36 | 43 | 2023 | India | Not specified | In silico | Tea leaves | Not stated | Camellia sinensis | Extracts |
The temporal distribution of studies indicates a steady increase in the use of network pharmacology and molecular docking in herbal medicine research presented in Figure 2. Early activity was limited, with only 2.78% of studies published in 201315 and 5.56% in 2016, reflecting the initial stage of adoption. A small increased appeared in 2020 (2.78%), followed by more noticeable growth in 2021 (8.33%), marking the beginning of wider implementation of computational methods in the field. A significant expansion occurred from 2022 onward, where publication frequencies reached 19.44% in 2022 and 13.89% in 2023.11 The highest proportion of studies was observed in 2024 (30.56%),19,11 and a substantial number continued to be published in 2025 (16.67%).4
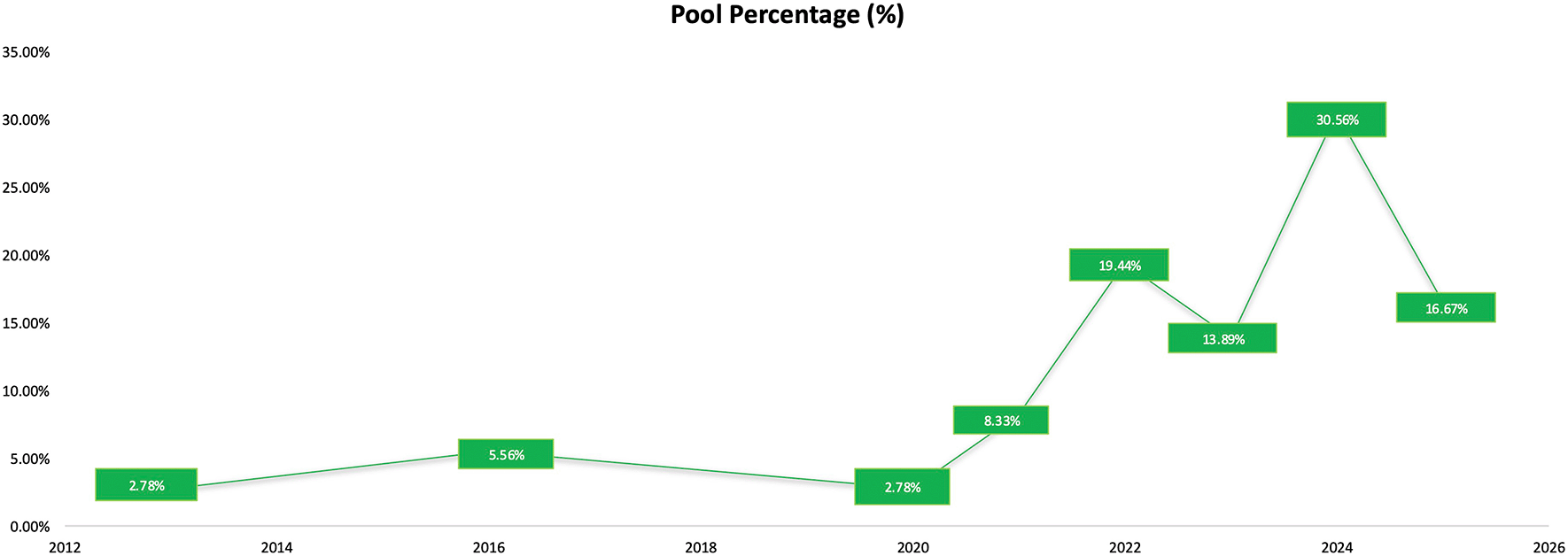
The temporal distribution shows studies were reported in 2013 (2.78%), 2016 (5.56%), 2020 (2.78%), 2021 (8.33%), 2022 (19.44%), 2023 (13.89%), 2024 (30.56%), and 2025 (16.67%).
The included studies utilized different computational databases to identify phytochemicals, predict molecular targets, and characterize disease-associated genes. These databases included herbal/phytochemical repositories, chemical-structure databases, target-prediction platforms, and disease–gene resources. Additional tools were used for ADME–toxicity screening and protein interaction analysis.
3.3.1 Herbal/phytochemical repositories
Across the included studies, TCMSP was the most frequently used herbal database, accounting for 52.9% of all phytochemical database citations as presented in Figure 3. This was followed by IMPPAT (17.6%) and TCM Database@Taiwan (11.8%),15 whereas HERB, ETCM, and NPASS were each used in 5.9% of studies. These findings suggest that most researchers rely on widely established Chinese herbal databases, with relatively lower utilization of newer or region-specific phytochemical repositories.
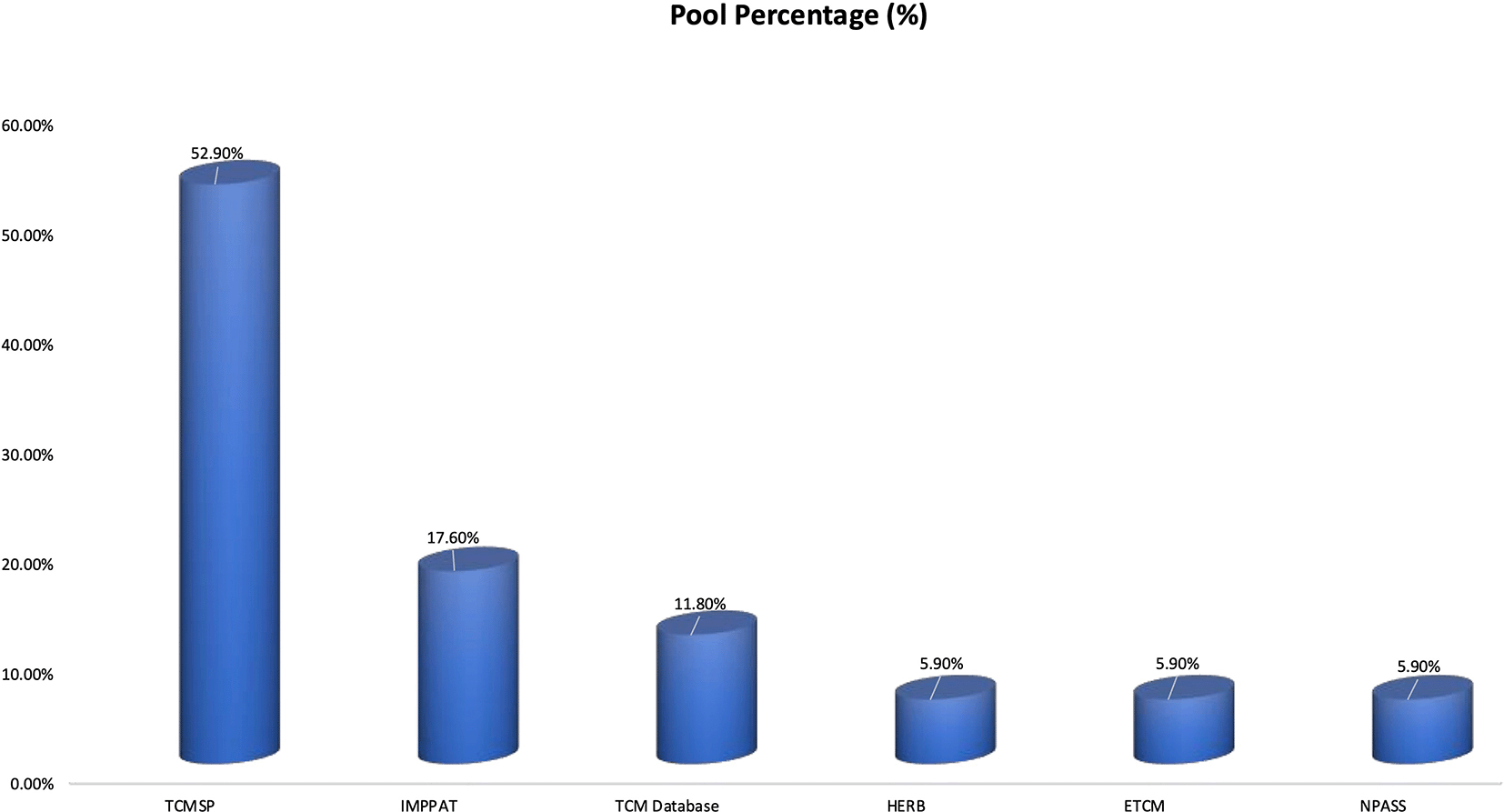
The figure illustrates the frequency of herbal database utilization across the reviewed literature (n = 17 mentions). TCMSP was the most commonly employed platform (52.9%), followed by IMPPAT (17.6%) and TCM Database@Taiwan (11.8%). HERB, ETCM, and NPASS were each used in 5.9% of studies.
3.3.2 Target-prediction platforms used in included studies
SwissTargetPrediction was reported in 36.36% of studies (n = 8), representing the most frequently used platform for target prediction ( Table 2). DIGEP-Pred, SEA (Similarity Ensemble Approach), and non-specified or model-based approaches were each reported in 9.09% of studies (n = 2 each), indicating moderate use across the included studies. PharmMapper, Way2Drug, SuperPred, ImaGEO, STITCH (used for target assignment), STRING (used for protein interaction-based target assignment), TTD, and PharmGKB were each reported in 4.55% of studies (n = 1 each).
| Target prediction platform | Frequency (n = 22) | Percentage (%) | Reference |
|---|---|---|---|
| SwissTargetPrediction | 8 | 36.36% | 16 |
| DIGEP-Pred | 2 | 9.09% | 27 |
| SEA | 2 | 9.09% | 26 |
| Not specified/model-based | 2 | 9.09% | 35 |
| PharmMapper | 1 | 4.55% | 18 |
| Way2Drug | 1 | 4.55% | 19 |
| SuperPred | 1 | 4.55% | 26 |
| ImaGEO | 1 | 4.55% | 12,16 |
| STITCH (target assignment) | 1 | 4.55% | 8 |
| STRING (target assignment) | 1 | 4.55% | 35 |
| TTD | 1 | 4.55% | 29 |
| PharmGKB | 1 | 4.55% | 17 |
The results show that SwissTargetPrediction was the most commonly employed platform (36.36%), followed by DIGEP-Pred, SEA, and non-specified computational approaches (each 9.09%). PharmMapper, Way2Drug, SuperPred, ImaGEO, STITCH, STRING, TTD, and PharmGKB were each used in 4.55% of studies, indicating selective adoption of specialized tools for structure-based prediction, transcriptomic inference, and protein-interaction mapping.
3.3.3 Chemical structure databases
Chemical structure databases were used to support compound identification and structural verification across the included studies as depicted in Figure 4. PubChem was the most frequently used resource (31.25%), followed by SwissADME (18.75%). Other tools including ChEMBL,22 admetSAR, ChemDraw,23 NIST, Dictionary of Natural Products, and CDRUG were each reported in 6.25% of cases. This distribution reflects a primary reliance on established public chemical repositories such as PubChem, with supplementary use of cheminformatics and property-prediction tools for compound validation.
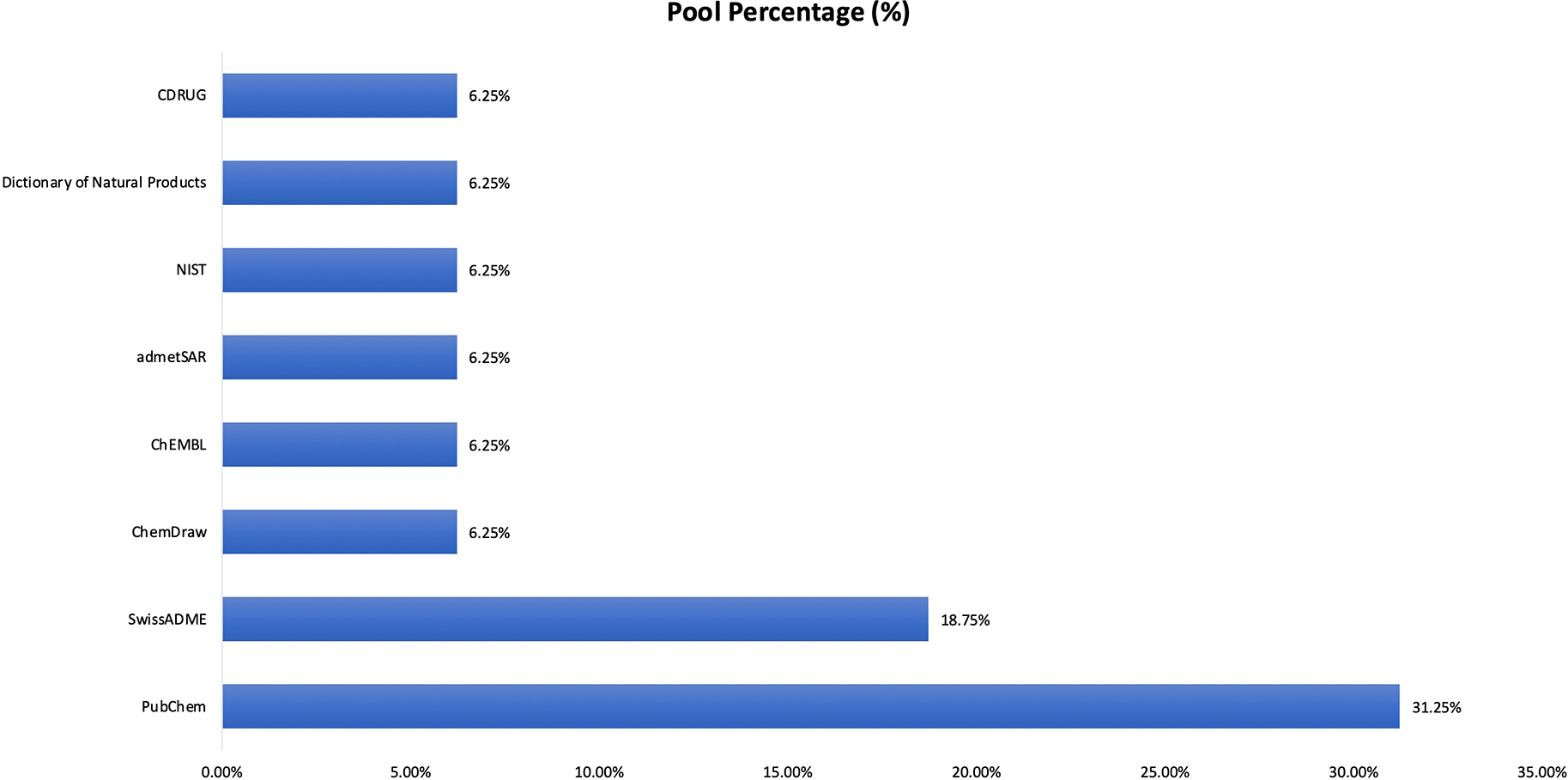
The result show that pubChem was most commonly employed (31.25%), followed by SwissADME (18.75%), with ChEMBL, admetSAR, ChemDraw, NIST, Dictionary of Natural Products, and CDRUG each used in 6.25% of studies.
3.3.4 Disease-gene annotation databases used across included studies
Disease-gene retrieval in the included studies that primary relied on GeneCards (44.44%), followed by OMIM and DisGeNET (22.22% each) as depicted by Figure 5. GEO, NCBI, and HPO were each utilized in 5.56% of studies. This pattern indicates a primary dependence on comprehensive disease-gene compendiums, particularly GeneCards,16 supplemented by clinically annotated resources (OMIM) and disease-association platforms (DisGeNET). Use of curated transcriptomic repositories (GEO)21 was less common, reflecting fewer studies integrating omics-driven disease gene mining.
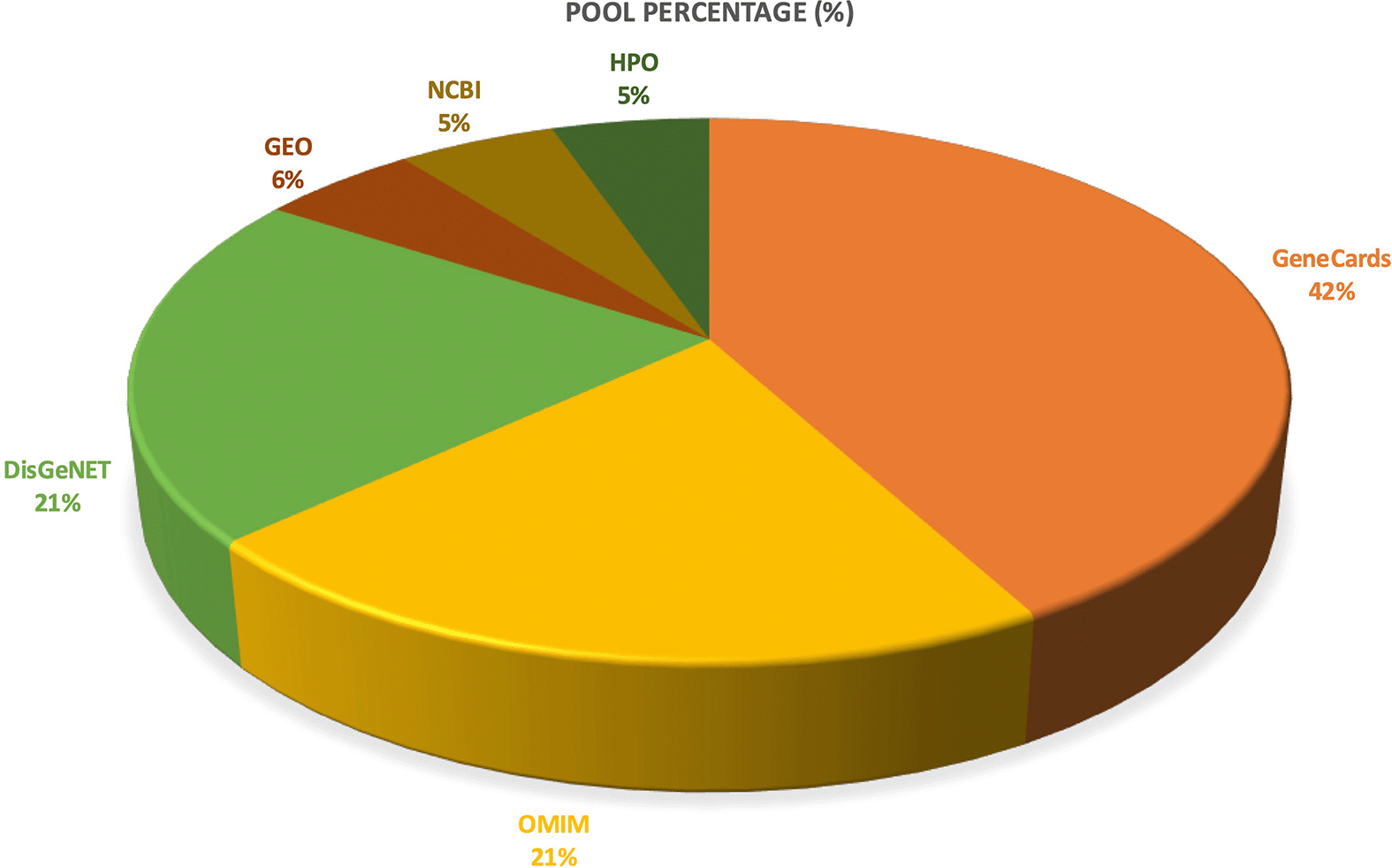
The figure results of the Disease-gene retrieval revealed GeneCards as the most frequently used disease-gene source (44.44%), followed by OMIM and DisGeNET (22.22% each). GEO, NCBI, and HPO accounted for 5.56% each, indicating selective incorporation of transcriptomic and phenotype-ontology sources.
3.3.5 Protein annotation and interaction databases used in the included studies
Across the included studies, protein annotation and interaction resources were used to support target validation and functional characterization as presented in Figure 6. UniProt/UniProtKB was the most frequently applied database (62.5%), indicating its central role in annotating protein targets and linking phytochemicals to biological functions. BindingDB appeared in 25.0% of studies, primarily for retrieving experimental protein ligand interaction data, while STITCH was used in 12.5% of studies for identifying chemical protein interaction networks. Together, these tools provided essential support for confirming target relevance and mapping molecular interaction profiles in network-pharmacology workflows.
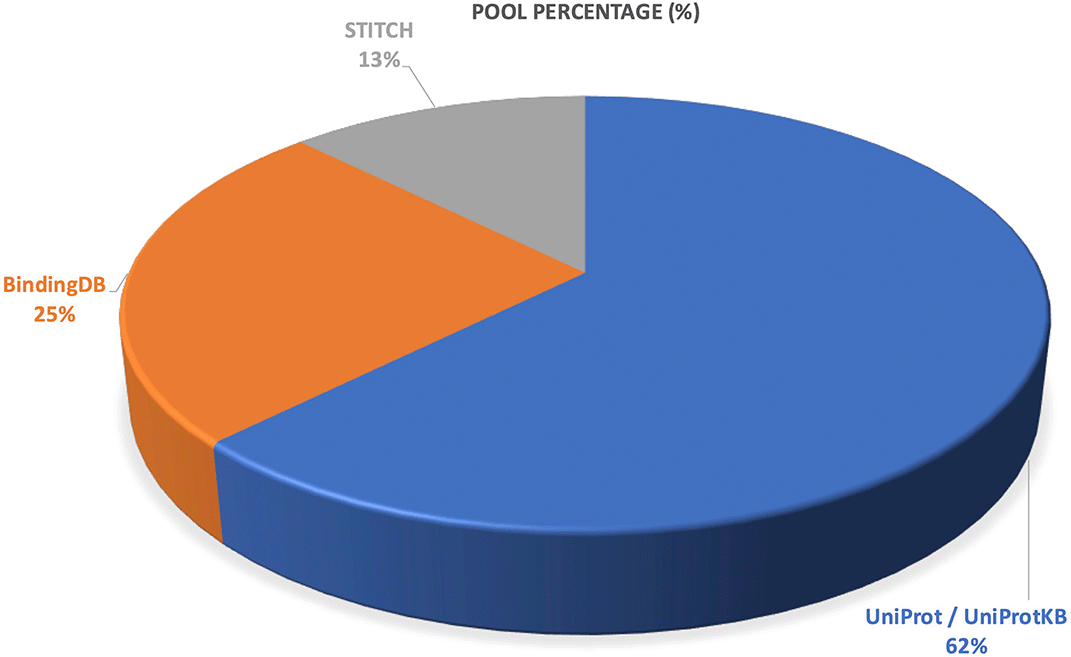
The results show that UniProt/UniProtKB was the most widely used platform, accounting for 62.5% of studies, followed by BindingDB (25.0%) and STITCH (12.5%). These resources were primarily applied for protein functional annotation, target confirmation, and mapping chemical–protein interaction networks within network-pharmacology pipelines.
3.3.6 Computational multi-database integrators
Among the included studies, some applied multi-database integration approaches that combine chemical, genomic, proteomic, and pathway data within a single workflow. These approaches were used to improve compound identification, increase confidence in target prediction, and support network construction. Reported platforms included HERB-linked compound and serum-metabolite integration frameworks, the Traditional Chinese Medicine Integrated Platform (TCMIP v2.0), and multi-omics pipelines integrating drug–target, protein–protein interaction, and pathway data (e.g., HERB, TCMIP v2.0), each reported once (12.5%). Other studies used combined database approaches for specific analyses. These included pathway enrichment (e.g., Metascape), compound validation using multiple chemical databases (e.g., PubChem, ChEMBL, Dictionary of Natural Products), and integration of experimental data (e.g., LC-MS/GC-MS) with disease–gene databases (e.g., DrugBank, OMIM, DisGeNET, GeneCards). One study also used a combination of a herbal database and viral-target literature to support antiviral drug discovery.
3.3.7 Literature-derived compound sources
Reported sources included literature-derived compound datasets integrated with filtering tools (e.g., PreADMET, Osiris Property Explorer) and manual literature searches combined with phytochemical and ADME databases (e.g., IMPPAT, SwissADME). Some studies cross-validated literature-reported compounds using target and disease databases (e.g., SwissTargetPrediction, GeneCards, DisGeNET). In several cases, studies developed in-house compound libraries based entirely on literature mining, while others used literature-supported compound lists alongside established herbal databases (e.g., TCM Database@Taiwan). Additional approaches included literature-based repurposing datasets for antiviral compounds (e.g., SARS, MERS, SARS-CoV-2) and disease-specific compound extraction from published studies (e.g., prostate cancer data). Overall, ten studies (12.5%) incorporated literature-derived phytochemical sources as part of their compound identification strategy.
Inflammatory and immune-mediated conditions accounted for 14.04% of studies, followed by oncology-related investigations. Cancer-focused studies included breast cancer (8.77%), lung cancer (5.26%), colon/colorectal cancer (3.51%), prostate cancer (3.51%), hepatocellular carcinoma (1.75%), cervical cancer (1.75%), and unspecified cancer types (10.53%). COVID-19-related studies accounted for 7.02% of the dataset. Neurological and psychiatric conditions included Alzheimer’s disease (1.75%), Parkinson’s disease (1.75%), and anxiety/depression (3.51%). Metabolic and cardiovascular conditions comprised diabetes (1.75%), hypertension (1.75%), vascular calcification (1.75%), and oxidative stress-related disorders (1.75%). Gastrointestinal immune disorders, including ulcerative colitis (3.51%) and Crohn’s disease (1.75%), were also reported ( Table 3).
| Condition | Frequency (n) | Percentage (%) | Reference |
|---|---|---|---|
| Inflammation/Anti-inflammatory | 8 | 14.04 | 14 |
| Cancer (unspecified) | 6 | 10.53 | 27 |
| Breast cancer | 5 | 8.77 | 37 |
| COVID-19 | 4 | 7.02 | 39 |
| Lung cancer | 3 | 5.26 | 19 |
| Colorectal/Colon cancer | 2 | 3.51 | 21 |
| Anxiety/Depression | 2 | 3.51 | 44 |
| CNS diseases (unspecified) | 2 | 3.51 | 15 |
| Hematologic diseases (unspecified) | 2 | 3.51 | 15 |
| Immune system diseases (unspecified) | 2 | 3.51 | 29 |
| Prostate cancer | 2 | 3.51 | 23 |
| Ulcerative colitis | 2 | 3.51 | 16 |
| Alzheimer’s disease | 1 | 1.75 | 40 |
| Cervical cancer | 1 | 1.75 | 26 |
| Crohn’s disease | 1 | 1.75 | 21 |
| Diabetes | 1 | 1.75 | 40 |
| Digestive diseases (unspecified) | 1 | 1.75 | 15 |
| Gout | 1 | 1.75 | 10 |
| Hepatocellular carcinoma | 1 | 1.75 | 32 |
| HIV-1 | 1 | 1.75 | 8,38 |
| Hypertension | 1 | 1.75 | 25 |
| Oxidative stress/Antioxidant | 1 | 1.75 | 34 |
| Parkinson’s disease | 1 | 1.75 | 33 |
| Vascular calcification | 1 | 1.75 | 20 |
| Infectious diseases (unspecified) | 1 | 1.75 | 8 |
| Lung cancer (SCLC) | 1 | 1.75 | 28 |
The results show that inflammatory and immune-mediated disorders were the most frequent focus (14.04%), followed by cancer-related investigations (breast cancer 8.77%, cancer-unspecified 10.53%, lung cancer 5.26%, colon and prostate cancer each 3.51%, and hepatocellular and cervical cancer each 1.75%). COVID-19 accounted for 7.02% of studies, while neurological disorders (Parkinson’s and Alzheimer’s disease), metabolic and cardiovascular conditions (diabetes, hypertension, vascular calcification), and gastrointestinal immune diseases (ulcerative colitis, Crohn’s disease) each represented 1.75–3.51% of the literature.
Across the included studies, Cytoscape emerged as the primary network-pharmacology platform, appearing in 63.89% of cases as presented in Table 4. Among these, versions v3.7.2 and v3.8.223 were most frequently reported (8.33% each), while other versions including v3.7.0,24 v3.10, v3.9.0, v3.9.1, v3.7.1, and v3.2 were each identified in 2.78% of studies. Nevertheless, version information was omitted in 30.56% of Cytoscape-using studies, indicating incomplete reporting of software details. Beyond Cytoscape, STRING23 was the next most commonly used platform (22.22%), followed by AutoDock-based pipelines (11.11%) and STITCH (5.56%). Less frequently adopted tools included GEPIA2, KEGG/GO16,20 enrichment platforms, Python NetworkX, and machine-learning-driven approaches (each 2.78%). Additionally, 19.44% of studies applied network-pharmacology workflows without specifying the software used, highlighting variability and reporting gaps in computational methodology.
| Software/Platform | Frequency (n) | Percentage (%) | |
|---|---|---|---|
| Cytoscape v3.7.2 | 3 | 8.33% | 18 |
| Cytoscape v3.8.2 | 3 | 8.33% | 23 |
| Cytoscape v3.7.0 | 1 | 2.78% | 24 |
| Cytoscape v3.10 | 1 | 2.78% | 27 |
| Cytoscape v3.9.0 | 1 | 2.78% | 32 |
| Cytoscape v3.9.1 | 1 | 2.78% | 30 |
| Cytoscape v3.7.1 | 1 | 2.78% | 33 |
| Cytoscape v3.2 | 1 | 2.78% | 7 |
| Cytoscape (version not specified) | 11 | 30.56% | 17,21 |
| STRING | 8 | 22.22% | 23 |
| AutoDock/PyRx docking workflow | 4 | 11.11% | 17 |
| STITCH | 2 | 5.56% | 18 |
| GEPIA2 | 1 | 2.78% | 19 |
| KEGG/GO enrichment platforms | 1 | 2.78% | 16 |
| Python NetworkX | 1 | 2.78% | 8 |
| Machine-learning pipeline (SVM/MLP/RF) | 1 | 2.78% | 40 |
| Unspecified network-pharmacology tools | 7 | 19.44% | 42 |
The results indicate different computational workflows, with multiple versions of Cytoscape widely used (8.33% each for v3.7.2 and v3.8.2; 30.56% unspecified). STRING was applied in 22.22% of studies, and AutoDock-based pipelines in 11.11%. STITCH and other tools including GEPIA2, KEGG/GO enrichment tools, Python NetworkX,8 and machine-learning models each appeared in 2.78% of studies. Additionally, 19.44% of studies referenced network-pharmacology pipelines without specifying software, highlighting reporting variability.
The distribution of pathway and enrichment tools shows that KEGG Pathway Analysis accounted for 37.88%, while Gene Ontology (GO) accounted for 28.79% and DAVID for 16.67%. Reactome and BioCarta were each reported at 3.03%. ShinyGO, KOBAS, BBID, WikiPathways, GOBP (GO Biological Process), Comparative Toxicogenomics Database network enrichment, and enrichment factor (EF) analysis were each reported at 1.52%, indicating that these tools were applied individually across different studies ( Table 5).
| Tool/Platform | Frequencies (n) | Percentages (%) | Reference |
|---|---|---|---|
| KEGG Pathway Analysis | 25 | 37.88 | 17,20 |
| Gene Ontology (GO) | 19 | 28.79 | 17 |
| DAVID | 11 | 16.67 | 18 |
| Reactome | 2 | 3.03 | 40 |
| BIOCARTA | 2 | 3.03 | 29 |
| ShinyGO | 1 | 1.52 | 30 |
| KOBAS | 1 | 1.52 | 33 |
| BBID | 1 | 1.52 | 35 |
| WikiPathways | 1 | 1.52 | 40 |
| GOBP (GO Biological Process) | 1 | 1.52 | 41 |
| C–T–D network enrichment | 1 | 1.52 | 8 |
| Enrichment factor (EF) analysis | 1 | 1.52 | 8 |
Distribution of pathway and enrichment analysis tools used across included studies. The results show that KEGG (37.88%) and Gene Ontology (28.79%) were the most frequently applied enrichment resources, followed by DAVID (16.67%). Other tools, including Reactome, BIOCARTA, ShinyGO, KOBAS, and WikiPathways, accounted for smaller shares (≤3.03%), demonstrating limited adoption beyond core KEGG- and GO-based platforms.
The analysis reveals a concentration of research efforts on key molecular regulators implicated in cancer progression, inflammation, cell survival, apoptosis, and angiogenesis as presented in Table 6. The most frequently investigated targets included ESR1 (27.78%), EGFR (25.00%),20 and AKT1 (22.22%), reflecting strong emphasis on hormone-dependent and growth-factor-mediated signaling pathways.23 Inflammation-related proteins such as TNF (19.44%), IL6 (11.11%), and IL1B (8.33%),17 alongside apoptosis-linked mediators (CASP3 and PTGS2, each 16.67%) were also prominently represented. Similarly, tumor suppressors and stress-response proteins (TP53 and HSP90AA1, each 13.89%) and angiogenesis and matrix-remodeling factors (VEGFA and MMP9, each 13.89%) appeared consistently across studies.
| Protein Target | Biological Role Category | Frequency (n) | Percentage (%) | Reference |
|---|---|---|---|---|
| ESR1 | Hormonal/Cancer signaling | 10 | 27.78% | 22 |
| EGFR | Growth factor/Cancer signaling | 9 | 25.00% | 20 |
| AKT1 | Cell survival/Metabolism | 8 | 22.22% | 23 |
| TNF | Inflammation/Immune regulation | 7 | 19.44% | 21 |
| CASP3 | Apoptosis execution | 6 | 16.67% | 23 |
| PTGS2 (COX-2) | Inflammation/Pro-tumorigenic | 6 | 16.67% | 16 |
| TP53 | Tumor suppressor/DNA repair | 5 | 13.89% | 27 |
| STAT3 | Immune & growth signaling | 5 | 13.89% | 24 |
| HSP90AA1 | Heat-shock stress response/Cancer | 5 | 13.89% | 20 |
| MMP9 | Extracellular matrix remodeling | 5 | 13.89% | 30 |
| VEGFA | Angiogenesis | 5 | 13.89% | 27 |
| MAPK1 | MAPK pathway signaling | 4 | 11.11% | 25 |
| IL6 | Cytokine signaling/Inflammation | 4 | 11.11% | 17 |
| CTNNB1 | Wnt/β-catenin signaling | 3 | 8.33% | 37 |
| IL1B | Cytokine signaling | 3 | 8.33% | 16 |
Overall, these patterns highlight a research emphasis on interconnected oncogenic and immuno-inflammatory pathways, demonstrating that herbal-network-pharmacology investigations predominantly target biologically relevant hubs associated with cancer, metabolic disorders, and inflammation-related diseases.
The results show that ESR1 (27.78%), EGFR (25%), and AKT1 (22.22%) were the most frequently investigated proteins, followed by inflammation-related factors such as TNF (19.44%) and CASP3/PTGS2 (16.67% each). Targets predominantly reflected pathways of cancer progression, inflammatory signaling, and metabolic regulation.
Across the included studies, phytochemical docking was more prevalence with focused on flavonoid compounds, with quercetin (25%),24 kaempferol (13.9%),16 apigenin (11.1%), and luteolin (8.3%) being the most frequently evaluated ligands as presented in Table 7. Phytosterols such as β-sitosterol (8.3%) and polyphenols including gallic acid (5.6%) and EGCG (5.6%) also appeared consistently. The remaining compounds (75%) were evaluated in single studies, indicating ongoing screening of diverse herbal metabolites spanning alkaloids, terpenoids, lignans, catechins, and phenolic acids.
| Phytochemical | Frequency (n) | Percentage (%) | Reference |
|---|---|---|---|
| Quercetin | 9 | 25.0% | 24 |
| Kaempferol | 5 | 13.9% | 16 |
| Apigenin | 4 | 11.1% | 32 |
| Luteolin | 3 | 8.3% | 34 |
| β-Sitosterol | 3 | 8.3% | 23 |
| Epigallocatechin gallate (EGCG) | 2 | 5.6% | 43 |
| Gallic acid | 2 | 5.6% | 37 |
| Emodin | 1 | 2.8% | 13 |
| Genistein | 1 | 2.8% | 13 |
| Naringenin | 1 | 2.8% | 24 |
| Isovitexin | 1 | 2.8% | 35 |
| Pipercine/Piperine | 1 | 2.8% | 37 |
| Resveratrol | 1 | 2.8% | 37 |
| Allicin/Diallyl trisulfide | 1 | 2.8% | 34 |
| Silibinin | 1 | 2.8% | 29 |
| Ecdysterone/20-HE | 1 | 2.8% | 23 |
| Others (unique single-study phytochemicals) | 27 | 75.0% | 16 |
Across the included studies, flavonoids dominated docking analyses, with quercetin (25%), kaempferol (13.9%), apigenin (11.1%), and luteolin (8.3%) being the most frequently evaluated compounds. Other commonly utilized phytochemicals included β-sitosterol (8.3%), gallic acid (5.6%), and EGCG (5.6%). Alkaloids, phenolic acids, steroidal compounds, and saponins appeared less frequently, typically in disease-specific screening contexts.
The distribution of reported binding affinities across the included studies shows that values ≤ −9.0 kcal/mol accounted for 19.4%, while values between −8.0 and −8.9 accounted for 16.7%. Affinity ranges of −7.0 to −7.9, −6.0 to −6.9, and −5.0 to −5.9 each accounted for 13.9%. Binding affinities greater than −5.0 accounted for 5.6%. Studies reporting mixed affinity ranges across −3 to −11.6 accounted for 8.3%, while unclear or unreported values also accounted for 8.3% ( Table 8).
| Binding Affinity Range (kcal/mol) | Frequency (n) | Percentage (%) | Reference |
|---|---|---|---|
| ≤ −9.0 (very strong binding) | 7 | 19.4% | 13 |
| −8.0 to −8.9 (strong binding) | 6 | 16.7% | 22 |
| −7.0 to −7.9 (moderately strong) | 5 | 13.9% | 32 |
| −6.0 to −6.9 (moderate affinity) | 5 | 13.9% | 24 |
| −5.0 to −5.9 (weak-to-moderate) | 5 | 13.9% | 20 |
| > −5.0/minimal affinity | 2 | 5.6% | 19 |
| Mixed ranges across all categories (broad −3 to −11.6) | 3 | 8.3% | 16 |
| Not reported/unclear | 3 | 8.3% | 35 |
Across the included studies (n = 36), the majority of phytochemicals demonstrated strong binding affinity, with 19.4% reporting very strong docking scores (≤ −9.0 kcal/mol) and 16.7% showing strong affinities (−8.0 to −8.9 kcal/mol). Moderate binding was observed in ~28% of studies, while weak/variable affinity (> −5 kcal/mol) was uncommon. Approximately 8.3% of studies did not specify binding values.
The analysis showed that PI3K–Akt signaling accounted (16.90%), followed by MAPK/ERK (14.08%)24 and NF-κB signaling (12.68%).21 Immune and cytokine-related pathways, including TNF, IL-17, and TLR cascades, represented 11.27% of reported pathways. Other reported mechanisms included Wnt/β-catenin signaling (7.75%), apoptosis pathways (7.04%), p53-mediated DNA-damage response (6.34%), and VEGF-linked angiogenesis pathways (6.34%). Pathways associated with oxidative stress (AGE–RAGE; 5.63%) and hypoxia response (HIF-1; 4.93%) were also commonly evaluated. In contrast, neurotransmission-related pathways (4.23%) and antiviral signaling mechanisms (2.82%) were less frequently reported ( Figure 7).
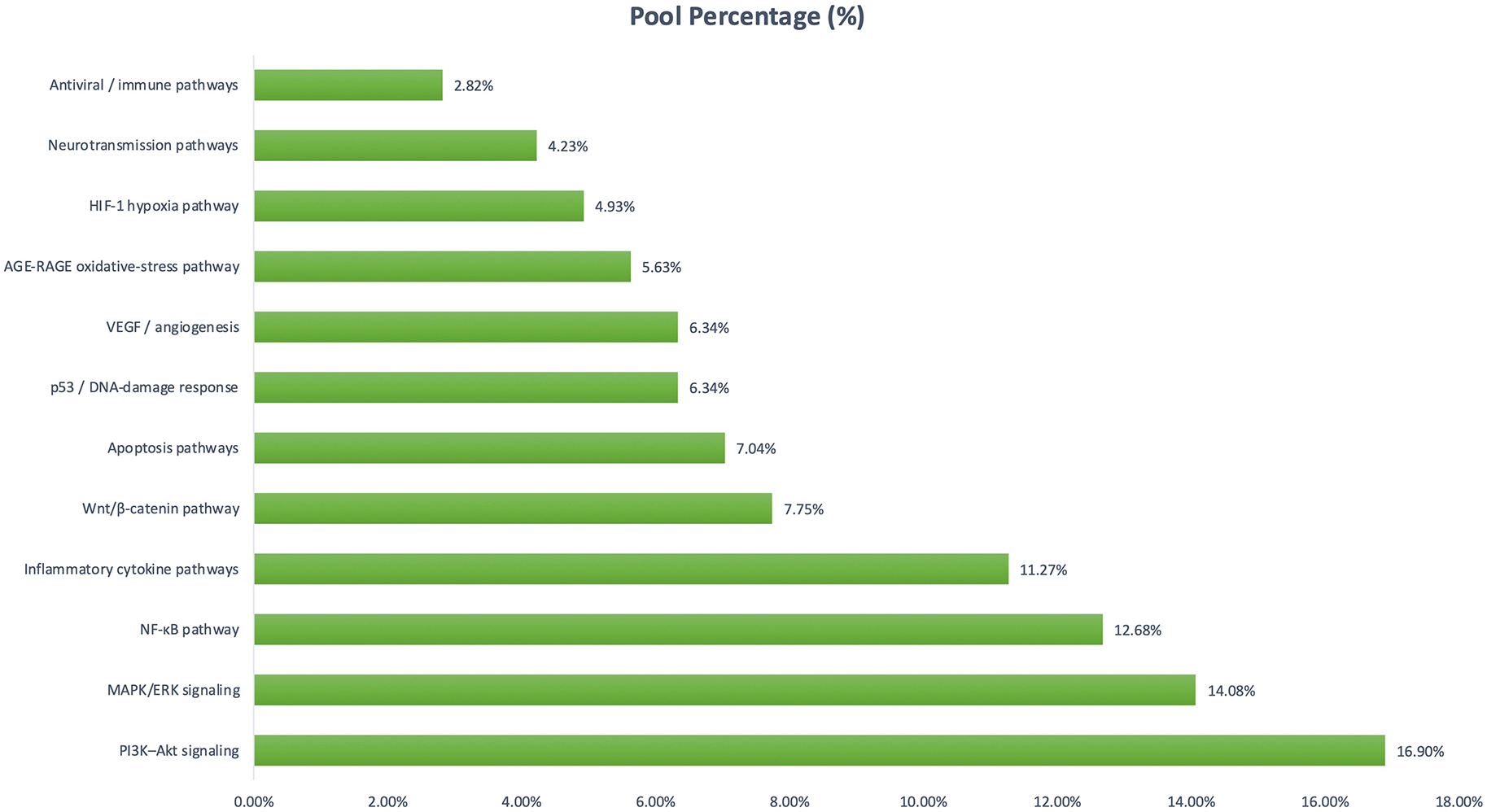
Distribution of molecular signaling pathways investigated across included studies based on total pathway mentions (N = 142). PI3K–Akt (16.90%), MAPK/ERK (14.08%), and NF-κB (12.68%) pathways accounted for the largest proportions, followed by inflammatory cytokine signaling (11.27%), Wnt/β-catenin (7.75%), apoptosis regulation (7.04%), and p53 and VEGF-related pathways (6.34% each). Oxidative stress (5.63%) and hypoxia responses (4.93%) were moderately represented, while neurotransmission (4.23%) and antiviral signaling (2.82%) were least frequently reported.
Across the included studies, ADMET and drug-likeness screening was commonly integrated into computational herbal research workflows as summarized in Table 9. SwissADME19 and TCMSP (OB & DL) were the most frequently used tools, each reported in 17.14% of studies. Lipinski’s rule of five was applied in 14.29% of the studies, highlighting consistent screening for oral bioavailability and small-molecule drug-likeness. Other predictive platforms such as admetSAR (8.57%), QikProp (5.71%), ProTox-II (2.86%), and pkCSM (2.86%) were used less frequently, suggesting selective adoption of advanced pharmacokinetic and toxicity profiling tools. DFT-based stability screening and MMGBSA/MDS simulations appeared in isolated studies (2.86% each), indicating emerging integration of quantum and dynamic computational methods. Notably, 28.57% of studies did not report performing ADMET evaluation, underscoring variability in reporting rigor across the literature.
| ADMET/Drug-likeness tool | Frequency (n) | Percentage (%) | Reference |
|---|---|---|---|
| SwissADME | 6 | 17.14% | 27 |
| TCMSP ADME (OB & DL) | 6 | 17.14% | 24 |
| Lipinski’s Rule of Five | 5 | 14.29% | 19 |
| admetSAR | 3 | 8.57% | 25 |
| QikProp (Schrödinger) | 2 | 5.71% | 29 |
| ProTox-II | 1 | 2.86% | 25 |
| pkCSM | 1 | 2.86% | 25 |
| PreADMET + Osiris | 1 | 2.86% | 30 |
| Way2Drug/PASS (toxicity) | 1 | 2.86% | 26 |
| MMGBSA/MDS (RMSD/RMSF) | 1 | 2.86% | 27 |
| Density Functional Theory (DFT) | 1 | 2.86% | 22 |
| Not specified/not performed | 10 | 28.57% | 17 |
Distribution of ADMET and drug-likeness evaluation tools across included studies. SwissADME and TCMSP screening (each 17.14%) were the most commonly used approaches, followed by Lipinski’s criteria (14.29%) and admetSAR (8.57%). Advanced computational pharmacokinetic tools such as QikProp, pkCSM, and ProTox-II were used less frequently (2.86%–5.71%). A considerable proportion of studies (28.57%) did not specify an ADMET assessment tool.
Across the 36 included studies, 17 studies (47.22%) incorporated molecular dynamics (MD) simulations as part of their computational workflow, while 19 studies (52.78%) did not perform MD simulations as depicted by Figure 8. This indicates that although MD simulation has emerged as a valuable technique for validating ligand–protein interactions and assessing structural stability, its adoption remains moderate, with just under half of studies integrating MD into their network-pharmacology or molecular docking analyses. Studies that employed MD simulations typically ran simulations ranging from 10 ns to 300 ns, often accompanied by complementary analyses such as MM-GBSA binding free energy calculation, RMSD, RMSF, PCA, FEL, and DCCM, demonstrating increasing methodological rigor among advanced studies.
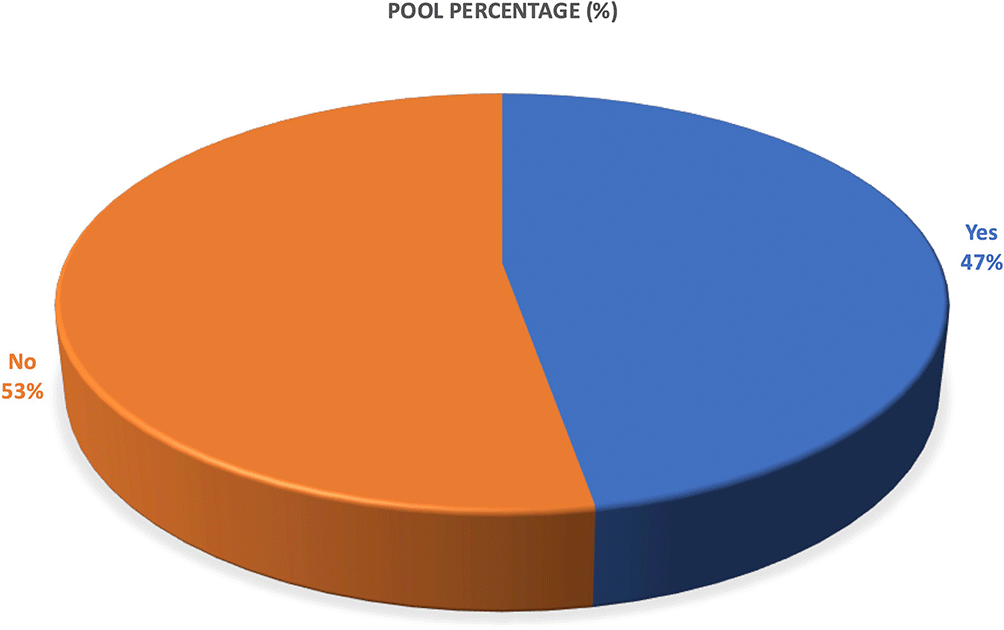
The results show that 47.22% (n = 17) of the studies conducted molecular dynamics simulations, while 52.78% (n = 19) did not incorporate MD procedures. This reflects a moderate adoption of MD simulation in natural-product-based computational drug-discovery studies.
Across the included studies, phytochemicals and herbal formulations demonstrated broad pharmacological activity mediated through multiple molecular pathways as summarized in Table 10. The most frequently reported mechanisms involved suppression of inflammatory mediators and immune signaling, particularly inhibition of NLRP3 inflammasome activation, NF-κB signalling, and pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6. A substantial proportion of studies also described anticancer effects driven by modulation of key oncogenic pathways including PI3K/Akt, MAPK,10,19 HIF-1α,20 and EGFR, alongside induction of apoptosis, cell-cycle arrest, and caspase activation. Neuroprotective effects were reported through attenuation of oxidative stress, stabilization of neuronal signaling, and inhibition of acetylcholinesterase activity. Additional mechanisms included metabolic and vascular regulation, antiviral activity (particularly SARS-CoV-2 and HIV-related targets), enhancement of intestinal barrier function, and modulation of endocrine signaling pathways. Overall, the results highlight a consistent trend toward multi-target and systems-level regulatory mechanisms, supporting the therapeutic relevance of network-guided phytochemical research.
| Mechanistic category | Key actions | Example targets/Pathways | Representative compounds | Reference |
|---|---|---|---|---|
| Anti-inflammatory and Immune Modulation | Suppresses inflammatory mediators, blocks inflammasomes, improves intestinal barrier | NLRP3, NF-κB, TNF-α, IL-6, IL-1β | Asiaticoside, Quercetin, Herbacetin, Costunolide derivatives | 23 |
| Anti-cancer/Anti-proliferative Effects | Inhibits tumor growth, induces apoptosis, blocks cell-cycle progression | EGFR, PI3K/Akt, MAPK, TP53, Cyclins (CDK1, Cyclin B1) | Acacetin, Sophoranone, Scutellarein, Episesamin | 22,23 |
| Cell Cycle Arrest & Apoptosis Induction | G2/M arrest, caspase activation, Bcl-2 downregulation | CDK1, CDC25A, PLK1, Caspase-3 | Biochanin A, Myrsininone A | 12,20 |
| Antioxidant and Oxidative-Stress Reduction | Enhances antioxidant enzymes, reduces ROS | Nrf2-related antioxidant defense | Ellagic Acid, EGCG, Kaempferol | 19 |
| Neuroprotective/Neuro-modulatory | Protects neurons, modulates neurotransmitters | NLRP3, acetylcholinesterase | Asiaticoside, AChE inhibitors | 17 |
| Vascular/Metabolic Regulation | Improves vascular function, lowers blood pressure, anti-diabetic enzyme inhibition | PI3K/Akt, AMPK, α-amylase, α-glucosidase | Allicin, Skimmin, Umbelliferone | 21 |
| Antiviral/Host-Immunity Support | Viral enzyme inhibition (COVID-19/SARS-CoV-2, HIV) | 3CLPro, HIV-PR, CXCR4, PD-1/PD-L1 | Emodin, Genistein | 40 |
| Gut Barrier & Microbiome Support | Enhances intestinal barrier integrity, regulates gut inflammation | RAGE/SLC6A14, STAT3 | JGD phytochemicals | 16 |
| Hormone/Endocrine Modulation | Modulates estrogen/androgen signaling | ESR1, CYP19A1, 5α-reductase | Isowighteone, Licochalcone A | 20 |
| Multi-target Network Regulation | Broad modulation of interconnected targets and pathways | PI3K/Akt, MAPK, HIF-1α, VEGF | Various poly-herbal extracts | 10 |
Natural compounds and herbal formulations demonstrated multi-target therapeutic actions across inflammation, cancer, metabolic regulation, neuroprotection, antiviral responses, gut barrier function, and hormonal pathways. Core mechanisms included suppression of pro-inflammatory mediators, modulation of PI3K/Akt and MAPK pathways, induction of apoptosis, inhibition of oxidative stress, and regulation of immune and neurotransmitter signaling. These findings support the systems-based therapeutic potential of phytochemicals in diverse chronic disease models.
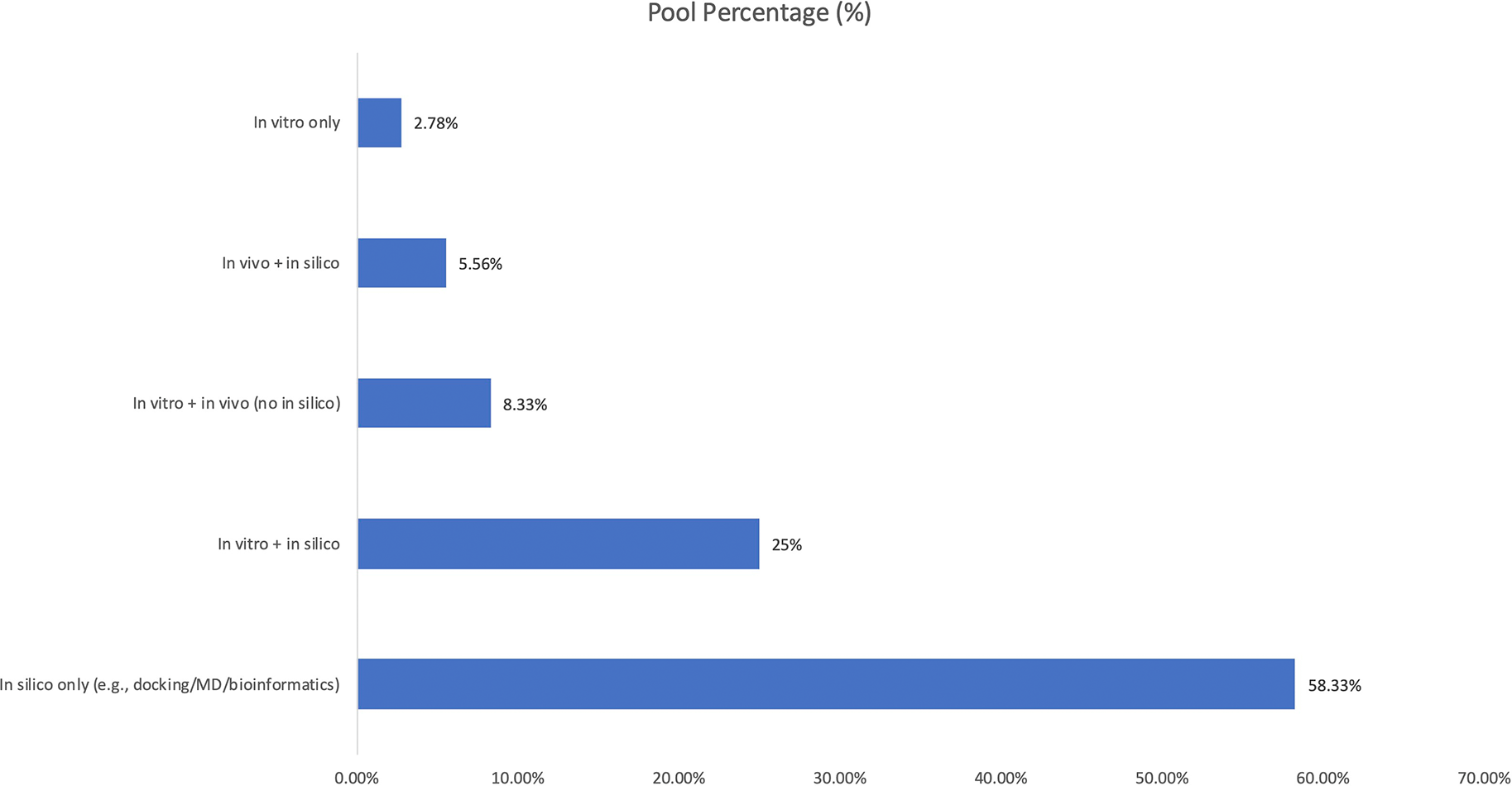
Across the 36 studies, in silico validation was the most frequent approach (58.33%), followed by combined in vitro + in silico designs (25.00%)16 as depicted in Figure 9. Fewer studies implemented laboratory animal validation,24 either with in vitro only (2.78%), in vivo + in silico (5.56%),23 or in vitro + in vivo without computational components (8.33%).10,19 No study used in vivo validation alone.
This systematic review assessed contemporary applications of network pharmacology combined with molecular docking in herbal medicine research from 2010 to 2025. The findings demonstrate that computational ethnopharmacology has progressed significantly during the last decade, particularly from 2020 onward. This represents global advances in artificial intelligence-driven drug discovery and increased scientific interest in natural products during the COVID-19 era.6 Notably, research activity increases significantly in 2024, highlighting that this field is growing and gaining momentum in mainstream biomedical science.9 Majority of the studies were originated from Asia, particularly China and India, accounting for over 80% of the total output. This geographic trend aligns with global patterns in which Traditional Chinese Medicine (TCM) and Ayurvedic herbal systems continue to drive innovation in plant-based computational pharmacology.4 The World Health Organization has similarly reported that East and South Asian nations lead in integrating computational tools into traditional medical systems.45 In contrast, regions rich in medicinal biodiversity such as Africa and South America remain under-represented. This represents a missed opportunity for global natural product discovery and emphasizing the need for wider international research participation and investment.46
Across the retrieved literature, there was a strong methodological preference for in silico-focused investigations, with over 60% of studies employing purely computational workflows. This confirms broader observations from recent reviews that computational strategies have become central to early-stage natural product screening.47 These strategies have been reported to be associated with improve target prediction, reduce laboratory cost, and minimize animal use.48 However, this trend simultaneously highlights an important translational gap. Few studies combined computational techniques with wet-lab validation, and even fewer extended to animal experimentation. These findings are consistent with prior analyses indicating insufficient experimental verification in network-pharmacology studies, which has raised concerns regarding reproducibility and biological relevance.5 Strengthening bench-to-bedside translation thus remains essential, particularly as network pharmacology continues to expand in clinical and drug-development frameworks.9
Furthermore, molecular dynamics simulation represented an emerging but not universal component of validation, being applied in nearly half of the studies. This adoption rate is higher than earlier reviews reported between 2020–2022, suggesting increasing methodological advancement.3 Nevertheless, variations in simulation duration, reporting formats, and analytic depth indicate that methodological standardization remains incomplete. Likewise, drug-likeness and ADMET filters, were widely acknowledged as necessary, were inconsistently applied, nearly one-third of studies failed to report such screening. This variability is consistent with international observations that computational phytomedicine lacks unified quality standards, reinforcing recent calls for harmonized methodological guidelines.2,49
Therapeutically, the studies addressed different disease systems. The strongest emphasis was observed in cancer, inflammation, immune dysregulation, metabolic disorders, infectious diseases, and neurological conditions. This pattern reflects the global disease burden and growing interest in systems-level natural-product therapeutics.46 Several mechanistic signatures appeared consistently across the literature.50 Phytochemicals frequently modulated PI3K-Akt, MAPK, NF-κB, IL-17, and apoptotic pathways, demonstrating their poly-target properties and supporting the rationale for network-based mechanistic inquiry. This aligns with international findings showing that natural compounds exhibit multi-pathway immunomodulatory and anti-cancer properties and tend to target central biological hubs rather than single pathways.1 In addition, the review identified a consistent focus on flavonoids like quercetin, kaempferol, luteolin, and apigenin molecules well documented in global research for broad anti-inflammatory, antioxidant, and anticancer activity.51,52 However, this reliance on widely studied phytochemicals also highlights a limitation where there is a risk that research pipelines may converge repeatedly on the same metabolites, restricting novel compound discovery. Future research should therefore balance investigation of established phytochemicals with systematic target on the lesser-studied plant metabolites, many of which remain pharmacologically uncharacterized.49 Another notable trend in this review is the frequent use of multi-herbal formulations, especially in studies originating from China and India. This contrasts with Western phytopharmacology, which traditionally prioritizes single-compound drug discovery.53 Multi-herbal computational analysis is particularly relevant for traditional medical systems that rely on poly-herbal synergy. However, it also presents challenges, as validating biological interactions within complex herbal mixtures requires advanced computational and experiment to avoid spurious associations.2
The review also revealed concentrated reliance on specific databases and platforms, including TCMSP, SwissTargetPrediction, STRING, UniProt, KEGG, and GeneCards. These tools indicate modern phytochemical research, dependence on primarily Asian phytochemical databases.14,38 Increased adoption of African and South American plant databases, integration of metabolomics, and broader utilization of global ethnobotanical repositories would improve heterogeneity and enhance natural-compound discovery pipelines.54 The relatively limited use of omics-driven disease-gene databases suggests that full integration of transcriptomic and proteomic signatures into herbal network pharmacology is still developing. As multi-omics approaches become more accessible, future studies should leverage proteogenomic data to improve disease-mechanism fidelity.55
Overall, this systematic review demonstrates that network pharmacology and molecular docking have become fundamental components of modern herbal research and are increasingly supported by advancement in computational tools. The convergence of AI, cheminformatics, multi-omics, and natural product pharmacology offers an important potential for drug discovery. However, achieving translational relevance will require stronger validation pipelines, expanded global participation, more consistent methodological standards, and increased discovery of novel plant-based molecules beyond the commonly investigated flavonoids. However, achieving meaningful translational impact requires addressing several gaps. Future studies should prioritize standard validation pipelines integrating in vitro, in vivo, and preferably clinical evidence to substantiate computational predictions. Methodological standardization, transparent reporting of ADMET and molecular dynamics analyses, and greater use of multi-omics data (e.g., proteomics, metabolomics, single-cell sequencing) will strengthen reproducibility and comparability across studies. Importantly, global participation must be broadened to include more contributions from Africa, Latin America, and other biodiversity-rich regions that remain under-represented despite rich traditional medical knowledge.
Despite providing a comprehensive synthesis of network pharmacology and molecular docking applications in herbal medicine, several limitations should be acknowledged. First, the review was restricted to studies published in English, which may have excluded relevant research, particularly from regions where herbal medicine innovations are frequently reported in local languages. Second, although three leading scientific databases were searched (PubMed, Scopus, and Web of Science), studies indexed in regional repositories or preprint platforms were not included, potentially limiting coverage of emerging work in low-resource research settings. Third, heterogeneity in reporting standards across studies posed challenges for direct comparison. Numerous articles did not clearly document experimental parameters such as solvent systems, extraction conditions, docking software versions, ADMET screening tools, or molecular dynamics protocols, leading to variability in methodological clarity and rigor. Fourth, a substantial proportion of included studies relied solely on in silico approaches without in vitro or in vivo validation, restricting the ability to fully assess biological relevance and translational readiness of computational predictions.
Additionally, the field lacks standardized quality-assessment frameworks for computational herbal pharmacology, and no formal risk-of-bias tool exists for network pharmacology studies, limiting the ability to systematically evaluate study quality. Publication bias is also possible, as studies reporting strong binding affinities or positive mechanistic findings are more likely to be published. Finally, the evolving nature of computational platforms and rapid developments in AI-driven drug-discovery tools mean that some techniques used in earlier studies may now be obsolete, potentially affecting comparability across the review period.
Based on the findings of this systematic review, future research and policy efforts should prioritize strengthening methodologies and translational potential in herbal pharmacology. Researchers are encouraged to adopt fully integrated computational pipelines that combine network pharmacology, molecular docking, ADMET profiling, and molecular-dynamics simulation before proceeding to in vitro and in vivo validation. Standardized reporting guidelines for computational herbal studies should also be developed to improve reproducibility and cross-study comparability. In addition, funding bodies and research institutions should support capacity building in computational drug discovery, particularly in low- and middle-income countries, to enhance global research equity. Collaborative frameworks between computational scientists, phytochemists, pharmacologists, and clinicians are essential to accelerate the translation of in silico findings into clinically relevant phytotherapeutics. Finally, fostering open-access databases for phytochemicals, target interactions, and experimental outcomes will facilitate transparency, innovation, and equitable access to scientific resources in natural-product-based drug discovery.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)