Keywords
Alzheimer’s disease, metabolic dysfunction, endothelial dysfunction, insulin resistance, oxidative stress, amyloid-beta, blood–brain barrier, neuroinflammation.
Alzheimer’s disease, metabolic dysfunction, endothelial dysfunction, insulin resistance, oxidative stress, amyloid-beta, blood–brain barrier, neuroinflammation.
The revised version of the manuscript has undergone restructuring as suggested by the reviewer to improve scientific depth, clarity, and mechanistic integration. The introduction was revised to emphasize Alzheimer’s disease (AD) as a multisystem disorder with major socioeconomic implications, while incorporating the role of chronic low-grade inflammation in linking metabolic syndrome, oxidative stress, and neurodegeneration. References were added throughout the Introduction to support all scientific statements. A dedicated methodology section describing the literature search strategy, inclusion and exclusion criteria, and study selection process was also incorporated.
Several new sections, figures, and tables were introduced to strengthen mechanistic discussion. A new a discussion on chronic low-grade inflammation and AD was added, together with updated schematic figures illustrating the relationship between metabolic dysfunction, endothelial injury, and neurodegeneration. Previous figures were redesigned to better represent insulin resistance as a systemic disorder affecting the brain and vascular endothelium. Additionally, new table summarizing AD and mechanisms of endothelial dysfunction was included to improve conceptual organization.
Section 4 was expanded to provide a more detailed explanation of how systemic and cerebral insulin resistance impair PI3K/Akt-mediated endothelial nitric oxide signaling and cerebral perfusion. Shared molecular pathways were reorganized into clearer mechanistic discussions focusing on oxidative stress, AGE–RAGE signaling, mitochondrial dysfunction, and inflammatory interactions but were not discussed separately to avoid redundancy.
The therapeutic section was also broadened through the inclusion of metformin, pioglitazone (PPAR-γ agonists), ceramide-targeting therapies, VEGF modulators, and sirtuin activators. Overall, the revised manuscript presents a more integrated metabolic–vascular–neurodegenerative framework for understanding AD pathogenesis and therapeutic intervention.
See the authors' detailed response to the review by Igbayilola Yusuff Dimeji
Alzheimer’s disease (AD) is the most common cause of dementia, accounting for approximately 60–70% of dementia cases globally. According to recent estimates, more than 55 million people worldwide are living with dementia, and this number is projected to exceed 139 million by 2050 due to population aging and increasing life expectancy (World Health Organization, 2021). AD represents one of the most significant public health, socioeconomic, and clinical challenges of the 21st century. The disease is characterized by progressive memory impairment, executive dysfunction, behavioral disturbances, and cognitive decline, leading to loss of independence, institutionalization, and increased mortality (Safiri S, et al., 2024). AD imposes economic and social costs on patients, caregivers, healthcare systems, and national economies, with global dementia-related healthcare expenditures estimated to exceed one trillion US dollars annually.
Traditionally, AD was primarily regarded as a neurodegenerative disorder characterized by extracellular deposition of amyloid-beta (Aβ) plaques and intracellular accumulation of hyperphosphorylated tau protein forming neurofibrillary tangles (Abdulkhaliq AA, et al., 2026). These pathological processes contribute to synaptic dysfunction, neuronal loss, and progressive cognitive deterioration. However, despite extensive therapeutic efforts targeting amyloid and tau pathology, clinical benefits have remained modest, suggesting that AD pathogenesis extends beyond classical neuronal abnormalities alone (Zhang J, 2025 et al., 2025).
Accumulating evidence suggest that AD as a complex multisystem disorder involving metabolic, vascular, inflammatory, and immunological dysfunction (Christodoulou RC, et al., 2026). Epidemiological studies have demonstrated that metabolic disorders such as type 2 diabetes mellitus, obesity, dyslipidemia, insulin resistance, and metabolic syndrome significantly increase the risk of AD development and progression (Chandrasekaran P, et al., 2024). These metabolic abnormalities disrupt cerebral glucose metabolism, impair insulin signaling pathways, alter mitochondrial energy production, and increase oxidative stress within neuronal tissues. Furthermore, chronic low-grade inflammation associated with metabolic syndrome promotes cytokine dysregulation, endothelial injury, and sustained neuroinflammatory activation (Safiri S, et al., 2024).
Furthermore, endothelial dysfunction and vascular impairment have emerged as important contributors to AD progression. Cerebral endothelial cells regulate cerebral blood flow, maintain blood–brain barrier (BBB) integrity, and preserve neuronal homeostasis (Kim S, et al., 2025). Endothelial dysfunction disrupts BBB permeability, impairs cerebral perfusion, reduces clearance of neurotoxic proteins such as Aβ, and facilitates inflammatory cell infiltration into the brain microenvironment. Chronic vascular injury also contributes to oxidative stress, ischemic damage, impaired neuronal metabolism, and synaptic dysfunction (Yue Q, et al., 2024).
Systemic metabolic abnormalities amplify oxidative stress, inflammation, and vascular dysfunction, while endothelial impairment further compromises cerebral glucose utilization and neuronal survival. This interaction creates a self-perpetuating cycle of metabolic stress, vascular injury, neuroinflammation, and progressive neurodegeneration (Christodoulou RC, et al., 2026). Studies have independently examined metabolic dysfunction or vascular impairment in AD, fewer reviews have comprehensively integrated these mechanisms with classical amyloid and tau pathology (Zhang, et al., 2024). Therefore, this review aims to synthesize current evidence regarding the mechanistic links between metabolic disorders, endothelial dysfunction, and Alzheimer’s disease progression.
This narrative review was conducted through a comprehensive literature search aimed at identifying studies related to metabolic dysfunction, endothelial dysfunction, and Alzheimer’s disease (AD). Electronic databases including PubMed/MEDLINE, Scopus, Web of Science, Google Scholar, and ScienceDirect were searched for relevant articles published between 2000 and 2026. Additional articles were identified through manual screening of reference lists from eligible studies. The search strategy combined Medical Subject Headings (MeSH) terms and free-text keywords related to AD pathogenesis and metabolic dysfunction. The main search terms included: “Alzheimer’s disease,” “metabolic dysfunction,” “insulin resistance,” “type 2 diabetes,” “endothelial dysfunction,” “oxidative stress,” “advanced glycation end-products,” “AGE-RAGE signaling,” “mitochondrial dysfunction,” “cerebral perfusion,” “blood–brain barrier,” “neuroinflammation,” “amyloid-beta,” “tau phosphorylation,” and “vascular pathology.” Boolean operators such as “AND” and “OR” were used to refine the search and improve retrieval of relevant studies.
Studies were included if they:
○ Investigated the relationship between metabolic dysfunction and Alzheimer’s disease.
○ Examined endothelial dysfunction, vascular injury, or cerebral perfusion abnormalities in AD.
○ Reported mechanistic, experimental, clinical, epidemiological, or therapeutic findings related to metabolic and vascular pathways in AD.
○ Were published in peer-reviewed journals in English.
○ Included animal studies, human studies, clinical trials, systematic reviews, and meta-analyses relevant to the topic.
Studies were excluded if they:
○ Were unrelated to Alzheimer’s disease or metabolic/endothelial dysfunction.
○ Focused exclusively on non-neurodegenerative disorders without relevance to AD mechanisms.
○ Were conference abstracts, editorials, commentaries, unpublished reports, or duplicate studies.
○ Lacked sufficient methodological or scientific detail.
○ Were published in languages other than English.
Titles and abstracts retrieved from the database search were screened for relevance to the review topic. Full-text articles of potentially eligible studies were then assessed based on the inclusion and exclusion criteria. Priority was given to recent studies, high-quality mechanistic investigations, clinical trials, systematic reviews, and landmark studies that provided substantial evidence on the metabolic–vascular–neurodegenerative axis in Alzheimer’s disease. Relevant data were synthesized narratively to provide an integrated understanding of the molecular mechanisms, pathological interactions, and emerging therapeutic strategies linking metabolic dysfunction and endothelial injury to AD progression.
Metabolic dysfunction plays a central role in the pathogenesis and progression of Alzheimer’s disease through interconnected mechanisms involving impaired glucose metabolism, oxidative stress, lipid dysregulation, and chronic inflammation, as illustrated in Figure 1.
The brain constitute about 2% of body weight, however it consumes approximately 20% of the body’s glucose-derived energy. This high metabolic demand makes it vulnerable to disruptions in glucose utilization. Insulin signaling in the brain play an important roles beyond glucose uptake. It regulates synaptic plasticity, neurotransmitter release, and neuronal survival through pathways such as phosphoinositide 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK). In AD, impaired insulin receptor function and downregulation of insulin receptor substrate proteins disrupt these signaling cascades (Seo et al., 2024). These dysfunctions have been associated with reduced neuronal glucose uptake and utilization, resulting to cerebral hypometabolism. Fluorodeoxyglucose positron emission tomography (FDG-PET) has demonstrated reduced glucose metabolism in the posterior cingulate, parietal, and temporal cortices, years before the onset of clinical symptoms (Martín-Saladich et al., 2025). Additionally, insulin resistance contributes directly to AD pathology with reduced insulin signaling decreases Akt activity, which normally inhibits glycogen synthase kinase-3β (GSK-3β). Overactivation of GSK-3β leads to tau hyperphosphorylation and the formation of neurofibrillary tangles. Similarly, impaired insulin signaling reduces clearance of amyloid-beta (Aβ) by downregulating insulin-degrading enzyme (IDE), allowing toxic peptide accumulation. This dual effect promotion of tau pathology and reduced Aβ clearance creates a pathological synergy that increase the risk of neurodegeneration. For example, obesity, metabolic syndrome, and type 2 diabetes (T2D) have been reported to increase the risk of cognitive decline, vascular dementia, and Alzheimer’s disease (J. E. Jun et al., 2026). Evidence from epidemiological, clinical, and basic research shows that neural dysfunction in T2D is driven by metabolic disturbances, inflammation, vascular injury, and oxidative stress (Jayaraman & Pike, 2014). Key modifiers include apolipoprotein E, a genetic risk factor, and low testosterone, an age-related endocrine change, both of which independently heighten Alzheimer’s risk and may synergistically worsen T2D-related neural damage. Moreover, hyperinsulinemia and elevated fasting glucose levels correlate with greater amyloid deposition and worse cognitive performance (Vaňková et al., 2023).
In insulin-resistant states, neurons experience impaired oxidative phosphorylation, leading to reduced ATP generation and insufficient energy supply to synapses as presented in Figure 1. This chronic energy deficit affects neuronal communication and resilience (Yuan et al., 2024). Impaired mitochondrial function increases electron leakage from the electron transport chain, particularly at complexes I and III. The resulting overproduction of reactive oxygen species (ROS) initiates a cascade of oxidative damage, targeting mitochondrial DNA, structural proteins, and membrane lipids. These changes compromise mitochondrial integrity, destabilize membranes, and promote the release of pro-apoptotic factors, ultimately driving neuronal apoptosis. Evidence from postmortem AD brains supports this mechanism, consistently showing altered mitochondrial morphology and reduced respiratory chain activity. Recent experimental work in mouse models of AD, mitochondrial redox stress measured with genetically encoded mt-roGFP sensors was elevated in neurons near amyloid plaques, with redox ratios rising by more than 30% compared to controls (Calvo-Rodriguez et al., 2024). Pharmacological interventions targeting mitochondrial calcium uptake or employing the antioxidant SS-31 normalized these redox signals and reduced plaque-associated neuritic damage, despite no reduction in plaque burden. Human imaging studies corroborate these findings with PET scans using [18F]BCPP-EF demonstrated reduced complex I availability in medial temporal regions of patients with mild AD, correlating with tau pathology and cognitive decline (Terada et al., 2021). This suggests that mitochondrial dysfunction is linked to tau-driven neurodegeneration. Genetic models also highlight causality with targeted disruption of the complex I subunit Ndufs4 in mice induced AD-like transcriptomic changes in the hippocampus, including alterations in synaptic and energy metabolism pathways (Gao et al., 2025). Treatment with a complex I modulator partially reversed these molecular signatures, indicating that mitochondrial dysfunction alone can initiate neurodegenerative cascades and is pharmacologically tractable (Yuan et al., 2024).
Abnormal lipid metabolism is one the major contributor to the pathogenesis of Alzheimer’s disease (AD). Dyslipidemia, characterized by increase low-density lipoprotein (LDL) cholesterol and reduced high-density lipoprotein (HDL), disrupts neuronal membrane composition and lipid raft stability. These lipid microdomains are important for synaptic signaling and amyloid precursor protein (APP) processing. High cholesterol levels favor amyloidogenic cleavage of APP by β- and γ-secretases, enhancing the generation of amyloid-beta (Aβ) peptides that aggregate into plaques. Apolipoprotein E (ApoE), particularly the ApoE4 isoform, plays a central role in cholesterol transport within the brain. Unlike ApoE2 or ApoE3, ApoE4 impairs lipid redistribution to neurons, prevent synaptic repair, and increase Aβ aggregation and deposition. Clinical evidence demonstrates that ApoE4 carriers have greater cortical amyloid burden and earlier onset of AD, accompanied by faster rates of cognitive decline. Hypercholesterolemia further stimulate vascular stiffness and reduces cerebral perfusion, compounding the combined effects of vascular and neurodegenerative injury (Raulin et al., 2022). In human cohorts, midlife hypercholesterolemia has been associated with increased late-life Aβ burden on PET imaging and faster progression to dementia, while statin exposure correlates with reduced AD risk, particularly among ApoE4 carriers (Panitch et al., 2021). ApoE4 knock-in mice subjected to high-fat/high-cholesterol diets exhibit increased Aβ deposition in cortical and hippocampal regions, worsened cerebral amyloid angiopathy, and reduced cerebral blood flow compared to ApoE3 controls (Ding et al., 2025). In vitro, increase membrane cholesterol in cultured neurons shifts APP processing toward the amyloidogenic pathway, while lipid raft disruption reduces Aβ generation. These findings emphasize dyslipidemia as amplifier of amyloid pathology and contributes to vascular dysfunction that synergizes with neurodegeneration (Rudajev & Novotny, 2022).
Chronic hyperglycemia, as observed in diabetes and metabolic syndrome increased non-enzymatic glycation of proteins and lipids, producing advanced glycation end products (AGEs). These AGEs form cross-links in extracellular matrix proteins, stiffening cerebral vasculature and impairing blood–brain barrier (BBB) integrity. AGEs act as potent pro-inflammatory mediators by binding to the receptor for advanced glycation end products (RAGE) on microglia, astrocytes, and endothelial cells. Engagement of the AGE–RAGE axis triggers NF-κB activation, which upregulates cytokines such as TNF-α, IL-1β, and IL-6. This chronic inflammatory environment enhances oxidative stress, promotes Aβ accumulation, and accelerates tau hyperphosphorylation. Additionally, Persistent low-grade inflammation contribute to the progression of Alzheimer’s disease (AD), particularly among individuals with obesity, metabolic syndrome, and type 2 diabetes mellitus (Ezkurdia A, et al., 2023). Excess adipose tissue promotes continuous release of inflammatory mediators such as TNF-α, IL-6, and IL-1β, creating a systemic inflammatory environment that extends beyond peripheral tissues and affects the brain. This prolonged inflammatory state alters vascular function, disrupts metabolic homeostasis, and increases susceptibility to neurodegeneration.
Furthermore, AGEs also accumulate with aging, meaning metabolic disorders intensify a process already driven by senescence, pushing the system toward neurodegeneration. Clinical data demonstrate that elevated circulating AGE levels correlate with poorer executive function and faster cognitive decline, underscoring their value as both biomarkers and mechanistic drivers of AD progression (Zoccali et al., 2025). In human studies, serum AGE indices and skin autofluorescence have been directly linked to cognitive impairment in diabetic and elderly cohorts (Mooldijk et al., 2024). In transgenic mouse models, crossing APP/PS1 lines with diabetic db/db mice elevates cortical AGE accumulation, microglial activation, and BBB disruption, while RAGE blockade reduces neuroinflammation and amyloid deposition (Jeong et al., 2022). At the cellular level, AGE-modified albumin exposure to microglia or endothelial cells activates NF-κB signaling and cytokine release, effects that are significantly attenuated when RAGE is silenced or pharmacologically inhibited. These findings highlight AGEs as a pivotal link between systemic hyperglycemia and central neuroinflammation in AD pathogenesis (Rezaee et al., 2024).
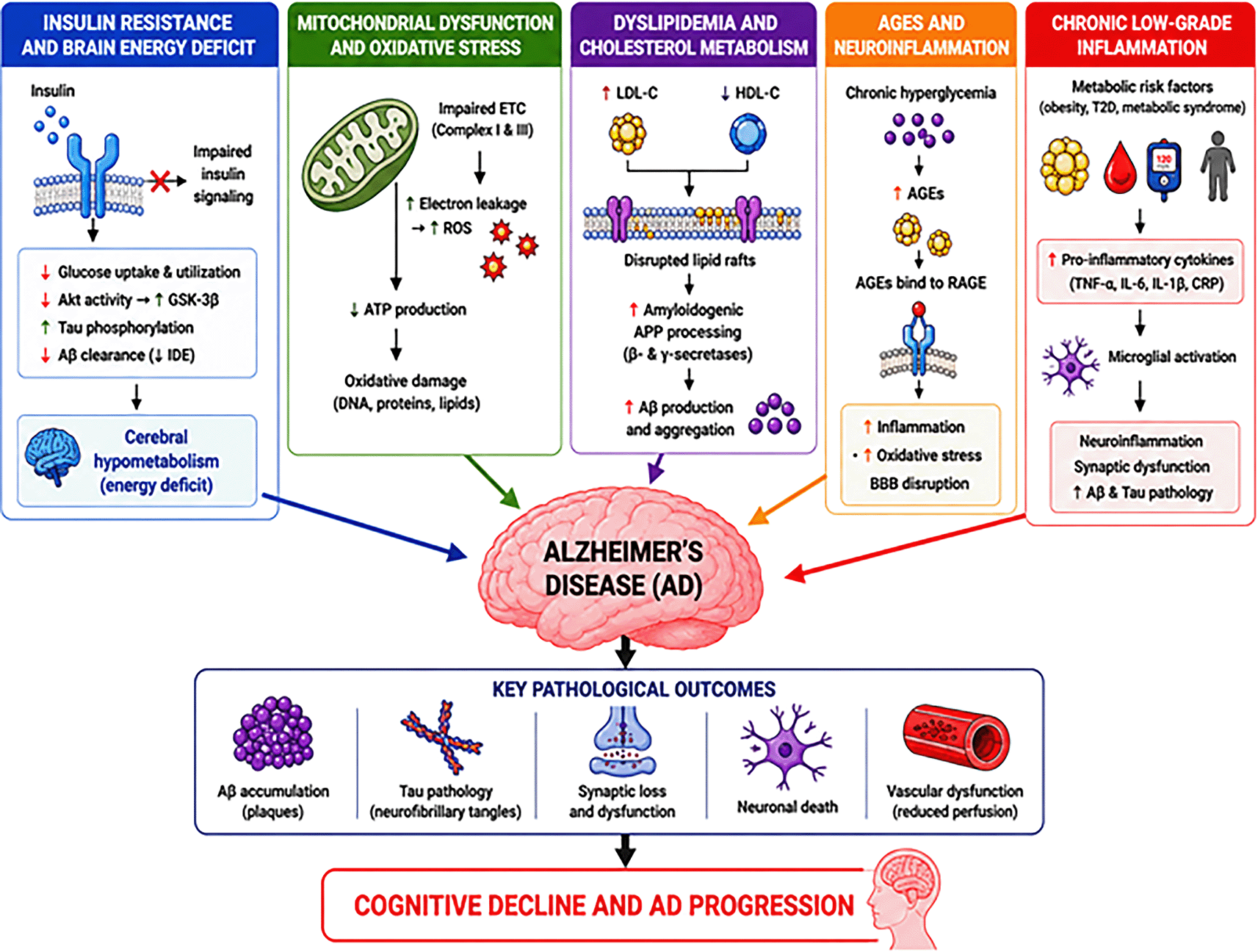
The figure illustrates how insulin resistance, mitochondrial dysfunction, dyslipidemia, advanced glycation end products (AGEs), and chronic low-grade inflammation contribute to amyloid-beta accumulation, tau pathology, oxidative stress, neuronal injury, vascular dysfunction, and cognitive decline in Alzheimer’s disease.
Endothelial dysfunction is a major contributor to Alzheimer’s disease progression through mechanisms involving blood–brain barrier disruption, impaired cerebral perfusion, oxidative stress, and vascular inflammation, as summarized in Table 1.
The vascular endothelium is fundamental to brain homeostasis, regulating blood flow, nutrient exchange, and blood–brain barrier (BBB) integrity. In the healthy brain, endothelial cells form tight junctions (claudins, occludins, ZO-1) that prevent uncontrolled leakage of plasma proteins and toxins. It allows regulated transport of essential molecules such as glucose through transporters like GLUT1 (Tomic et al., 2022). They also secrete vasoactive molecules including nitric oxide (NO) and prostacyclin, which maintain vascular tone and ensure adequate oxygen and nutrient delivery to regions of high neuronal activity. In individuals with chronic hypertension or diabetes, endothelial dysfunction results in reduced NO bioavailability, impaired vasodilation, and hypoperfusion of brain regions such as the hippocampus. This chronic hypoperfusion contributes to neuronal stress and increase the risk of cognitive decline. Similarly, reduced expression of GLUT1 in endothelial cells has been documented in AD patients, leading to energy deficits and impaired synaptic function (Yang et al., 2024). Endothelial cells express transporters such as low-density lipoprotein receptor–related protein 1 (LRP1), which mediates amyloid-beta (Aβ) clearance from the brain to circulation. When this system fails, Aβ accumulates in the brain parenchyma, forming plaques that drive AD pathology. A well-documented example is seen in APOE4 carriers, where impaired endothelial Aβ clearance accelerates plaque formation and correlates with early-onset cognitive decline (Darabi et al., 2025). A study using APP/PS1 mice crossed with endothelial nitric oxide synthase deficient (eNOS+/−) mice demonstrated that partial loss of eNOS worsens Alzheimer’s pathology. Compared with standard APP/PS1 mice, APP/PS1/eNOS+/− mice showed more severe spatial memory deficits, increased amyloid-beta (Aβ) plaque burden, upregulated BACE-1 (enhancing Aβ production), reduced insulin-degrading enzyme (limiting Aβ clearance), and increased microglial activation (Ahmed et al., 2022). A clinical study of 55 elderly participants showed that in MCI, the BBB was selectively more permeable to small molecules (e.g., water), but not to larger molecules like albumin. Increased permeability to water correlated with Alzheimer’s disease (AD) biomarkers (CSF Aβ, ptau) and predicted worse cognitive performance. In contrast, albumin permeability was associated with vascular risk factors, particularly hypercholesterolemia, but not AD pathology (Lin et al., 2021).
Endothelial nitric oxide synthase (eNOS) is a key regulator of cerebrovascular health, responsible for generating nitric oxide (NO), a vasodilator that maintains cerebral blood flow. It also responsible for regulating vascular tone, and modulates amyloid beta (Aβ) clearance across the blood–brain barrier (BBB). In Alzheimer’s disease (AD), eNOS activity is frequently impaired, leading to reduced NO bioavailability (Tran et al., 2022). This deficiency promotes vascular stiffness, endothelial dysfunction, and inadequate perfusion of neural tissue, creating an environment that increase neurodegeneration. The pathogenesis involves eNOS uncoupling, in which the enzyme shifts from producing NO to generating superoxide radicals due to deficiency of cofactors such as tetrahydrobiopterin (BH4) (Janaszak-Jasiecka et al., 2023). This impair vasodilatory capacity and amplifies oxidative stress through increased reactive oxygen species (ROS). Excess ROS further damages endothelial cells, oxidizes lipids, and enhances inflammatory signaling, creating a vicious cycle of vascular injury. In experimental studies, APP/PS1 transgenic mice with partial eNOS deficiency (APP/PS1/eNOS+/−) exhibited markedly higher Aβ deposition and more severe spatial memory deficits compared to APP/PS1 mice with intact eNOS. Mechanistic studies revealed upregulation of β-secretase (BACE-1), leading to greater Aβ production, and downregulation of insulin-degrading enzyme, reducing Aβ clearance (Ma et al., 2025). Increased microglial activation in these models further indicate the role of eNOS dysfunction in amplifying neuroinflammation and AD progression. Clinically, reduced eNOS activity has been associated with endothelial stiffness and impaired cerebral autoregulation in elderly individuals at risk for cognitive decline. Polymorphisms in the NOS3 gene (encoding eNOS) have also been linked to increased AD susceptibility, highlighting genetic contributions to endothelial dysfunction (An et al., 2021).
In Alzheimer’s disease (AD), systemic metabolic disorders including diabetes, obesity, and dyslipidemia increase the risk of BBB vulnerability by inducing chronic inflammation, oxidative stress, and endothelial injury (Dotiwala et al., 2023). This occurs as a results of disruption of tight junction proteins leading to increased vascular permeability. This process allows plasma proteins (e.g., fibrinogen, albumin) and peripheral immune cells to infiltrate the brain parenchyma. These infiltrates interact with amyloid precursor protein (APP) metabolism, accelerating amyloid-beta (Aβ) deposition and plaque formation. For example, fibrinogen binds directly to Aβ, enhancing aggregation and impairing microglial clearance, while albumin leakage alters osmotic balance and promotes local inflammation (Simões-Pires et al., 2025). For example, a study of 62 patients with mild cognitive impairment or dementia found that blood–brain barrier (BBB) permeability measured by MRI was associated with Alzheimer’s biomarkers (Moon Y et al., 2023). In amyloid-positive patients, higher BBB leakage was linked to lower Aβ40, altered Aβ42/40 ratio, reduced p-tau, and smaller hippocampal volume. In amyloid-negative patients, BBB leakage was associated with higher total tau (Moon et al., 2023). Similarly, pericyte injury, indicated by elevated soluble platelet-derived growth factor receptor-β (sPDGFRβ) in CSF, strongly correlated with BBB dysfunction and memory decline (Lv et al., 2023). Experimentally, in diabetic db/db mice, chronic hyperglycemia was shown to downregulate claudin-5 and occludin expression, causing BBB leakage and increased amyloid deposition in the hippocampus. In another study (Rom et al., 2020). Additionally, entry of thrombin and fibrinogen into the parenchyma activates microglia and astrocytes, promoting pro-inflammatory cytokine release (IL-1β, TNF-α) and worsening synaptic dysfunction. Plasma-derived albumin leaking into the brain binds to astrocytic TGF-β receptors, promoting excitotoxicity and epileptiform activity nphenomena observed in both rodent models and postmortem AD brains (Y. Chen et al., 2025).
Chronic metabolic stress, particularly in the context of type 2 diabetes, obesity, and hypertension, accelerates vascular aging by inducing endothelial senescence. Senescent endothelial cells lose their proliferative capacity and adopt a senescence-associated secretory phenotype (SASP), characterized by increased secretion of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α. This creates a self-perpetuating inflammatory loop within cerebral microvessels that promotes leukocyte adhesion, oxidative stress, and disruption of the neurovascular unit (Picos et al., 2025). Endothelial senescence is closely linked with cerebral small vessel disease (CSVD), which is highly prevalent in Alzheimer’s disease (AD) patients. CSVD contributes to white matter hyperintensities, lacunar infarcts, and microbleeds, all of which increased the risk of cognitive decline. Experimental models in ApoE-/- mice subjected to high-fat diets, markers of endothelial senescence (p16^INK4a, p21) are upregulated in cerebral arterioles, coinciding with increased blood–brain barrier (BBB) leakage and impaired cerebral perfusion (Fulop et al., 2018). Similarly, postmortem AD brain tissue demonstrates accumulation of senescent endothelial cells in cortical microvessels, correlating with both amyloid-β deposition and tau pathology (Gaikwad et al., 2023). Clinically, plasma biomarkers of vascular inflammation (e.g., soluble ICAM-1, VCAM-1, and circulating endothelial microparticles) have been associated with faster progression from mild cognitive impairment to AD. This suggests that endothelial senescence worsens vascular stiffness and hypoperfusion and also synergizes with amyloidogenic and tau-related pathways to accelerate neurodegeneration.
Metabolic and endothelial dysfunction reinforce each other in Alzheimer’s disease (AD). Insulin resistance, dyslipidemia, and hyperglycemia increase oxidative stress and AGEs, while impairing eNOS and cerebral perfusion. Endothelial injury induces by vascular stiffness, hypoperfusion, and BBB leakage increase the risk of neuronal energy deficits, amyloid accumulation, and inflammation. These interplay between metabolic dysfunction and endothelial dysfunction are detailed in the subsections that follow.
Insulin resistance, dyslipidemia, and hyperglycemia collectively damage endothelial cells by reducing nitric oxide (NO) production, increasing advanced glycation end-product (AGE) accumulation, and enhancing oxidative stress (Figure 2). Systemic and cerebral insulin resistance impair insulin-mediated activation of the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in endothelial cells. Under normal physiological conditions, insulin binding to endothelial insulin receptors activates insulin receptor substrate (IRS) proteins, which subsequently stimulate the PI3K/Akt pathway. Akt phosphorylates endothelial nitric oxide synthase (eNOS), promoting nitric oxide (NO) production that maintains vasodilation, vascular elasticity, and adequate cerebral blood flow (Yu, et al., 2026). In insulin-resistant states, impaired IRS signaling reduces PI3K/Akt activation and limits eNOS phosphorylation, resulting in decreased NO bioavailability, endothelial dysfunction, and vascular stiffness. Simultaneously, insulin resistance shifts signaling toward the mitogen-activated protein kinase (MAPK) pathway, which enhances endothelin-1 production, oxidative stress, inflammation, and vasoconstriction (Tang W, et al., 2026). These alterations compromise blood–brain barrier integrity, reduce glucose and oxygen delivery to neurons, impair amyloid-beta clearance, and promote chronic neuroinflammation, promoting Alzheimer’s disease progression (Moon et al., 2023). These changes result to reduced NO signaling and vascular stiffness impair cerebral blood flow and glucose delivery, depriving neurons of critical metabolic support. Additionally, BBB disruption allow the leakage of plasma proteins and infiltration of activated immune cells into the brain parenchyma. These process results to neuroinflammation, promoting microglial activation and oxidative stress, which in turn amplify Aβ production and tau hyperphosphorylation (Rom et al., 2020). The outcome is a vicious cycle with metabolic dysfunction increase vascular injury, vascular dysfunction promotes neuroinflammation, both increase the risk neuronal death. In APP/PS1/eNOS-deficient mice, partial loss of endothelial NO promote Aβ deposition, reduced clearance, and increase the risk spatial memory deficits compared with APP/PS1 mice alone. In high-fat diet rodent models, insulin resistance increased both tau phosphorylation and BBB permeability, directly linking systemic metabolism to neurovascular damage (An L et al., 2021). Similar findings have been reported in human studies in diabetic patients which show greater amyloid burden on PET imaging, more severe BBB permeability on MRI, and faster rates of cognitive decline compared to non-diabetics (Quenon et al., 2024).
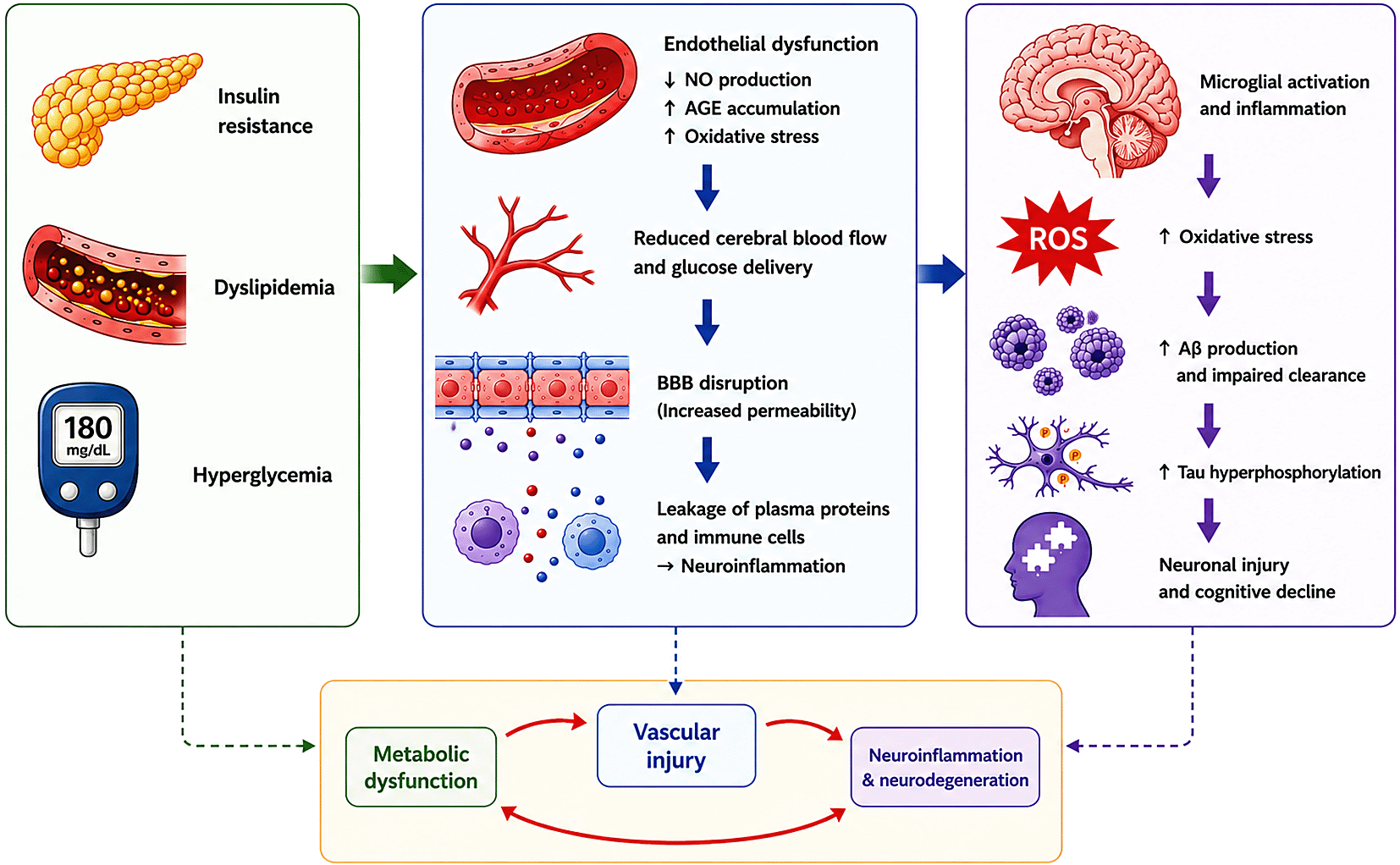
Insulin resistance, dyslipidemia, and hyperglycemia promote endothelial dysfunction through reduced nitric oxide (NO) production, increased advanced glycation end-product (AGE) accumulation, and oxidative stress. These vascular alterations impair cerebral blood flow, disrupt blood–brain barrier (BBB) integrity, and promote neuroinflammation. Progressive oxidative stress and inflammatory signaling enhance amyloid-beta (Aβ) accumulation, tau hyperphosphorylation, neuronal injury, and cognitive decline, forming a self-reinforcing cycle that accelerates AD progression.
Several molecular pathways connect metabolic dysfunction, endothelial injury, and neurodegeneration in Alzheimer’s disease (AD). Oxidative stress is one of the main shared mechanisms linking these processes. Insulin resistance, hyperglycemia, and mitochondrial dysfunction increase the production of reactive oxygen species (ROS) (Barone et al., 2021). In endothelial cells, ROS reduce nitric oxide (NO) availability by uncoupling endothelial nitric oxide synthase (eNOS), leading to vasoconstriction, vascular stiffness, and reduced cerebral perfusion as shown in Figure 3. In neurons, ROS damage mitochondrial DNA, proteins, and membrane lipids, impair synaptic signaling, and promote amyloid-beta (Aβ) accumulation (Barone et al., 2021). Experimental studies in streptozotocin-induced diabetic rats further showed that elevated ROS levels are associated with reduced cerebral blood flow and increased plaque deposition (Mohamed et al., 2022).
Another important shared pathway involves advanced glycation end-products (AGEs) and RAGE signaling. Chronic hyperglycemia increases AGE accumulation in vascular and neural tissues. AGEs bind to RAGE receptors expressed on endothelial cells, neurons, and glial cells, activating NF-κB–dependent inflammatory pathways and increasing cytokine production, including TNF-α, IL-1β, and IL-6 (Fulop et al., 2018). This inflammatory response promotes vascular stiffness, blood–brain barrier dysfunction, sustained microglial activation, and neuronal injury. AGE–RAGE signaling also enhances Aβ accumulation and tau hyperphosphorylation, directly linking metabolic imbalance to neurodegeneration. Clinical studies have shown that elevated AGE levels in diabetic patients are associated with faster cognitive decline, while inhibition of RAGE reduces neuroinflammation and plaque burden in AD mouse models (Ayoub et al., 2025).
Mitochondrial dysfunction further connects metabolic and vascular abnormalities. Impaired mitochondrial respiration reduces ATP production in both endothelial cells and neurons, weakening endothelial barrier integrity and neuronal energy metabolism. Damaged mitochondria also produce more ROS, which further increases oxidative stress and inflammation. As these pathways interact, oxidative stress accelerates AGE formation, AGEs increase inflammation and mitochondrial injury, and mitochondrial dysfunction generates additional ROS. This continuous interaction creates a self-reinforcing cycle that worsens endothelial dysfunction, reduces cerebral blood flow and glucose delivery, impairs Aβ clearance, and accelerates neuronal injury and cognitive decline in Alzheimer’s disease.
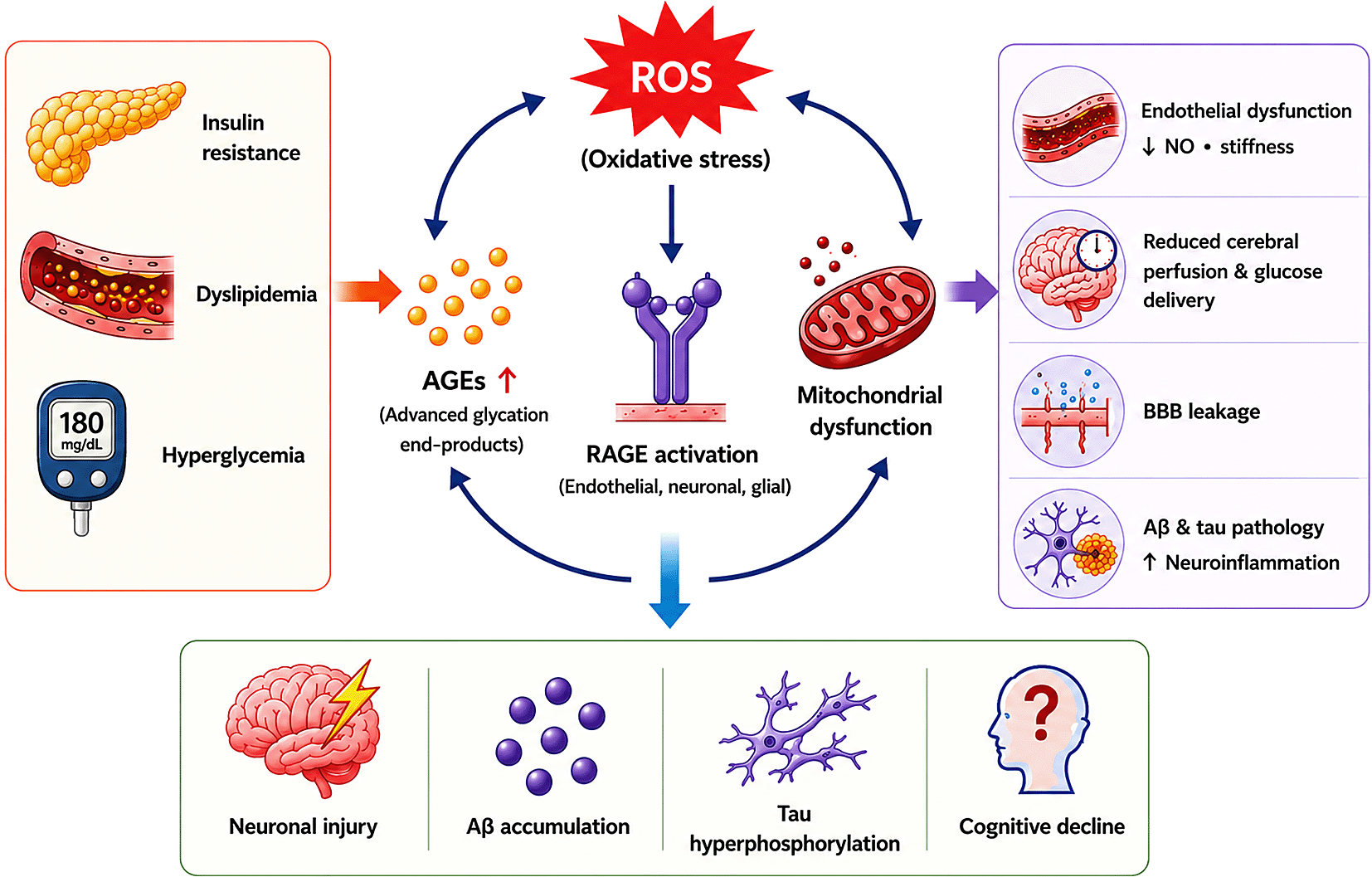
Schematic illustration showing the interconnected molecular pathway linking metabolic dysfunction to endothelial injury and neurodegeneration in Alzheimer’s disease (AD). Insulin resistance, dyslipidemia, and hyperglycemia promote oxidative stress, advanced glycation end-product (AGE) accumulation, and mitochondrial dysfunction. Reactive oxygen species (ROS) and AGE–RAGE signaling contribute to endothelial dysfunction, reduced cerebral perfusion, blood–brain barrier (BBB) leakage, neuroinflammation, amyloid-beta (Aβ) accumulation, and tau hyperphosphorylation, collectively driving neuronal injury and cognitive decline.
The recognition that Alzheimer’s disease (AD) emerges from the convergence of metabolic dysfunction and vascular pathology has broadened the scope of therapeutic exploration. Novel strategies increasingly focus on insulin signaling, endothelial repair, inflammation control, mitochondrial stabilization, and biomarker-driven personalization. Together, these innovations suggest a multi-targeted approach beyond the classical amyloid- and tau-centric model as summarised in Table 2.
The table summarizes major therapeutic categories with mechanistic relevance to Alzheimer’s disease, focusing on metabolic regulation, endothelial protection, and multi-target interventions. Representative treatments, molecular pathways, clinical advantages, and potential limitations are provided to highlight the mechanistic rationale behind each approach and current translational considerations. Key pathways include PI3K/Akt, AMPK, GSK-3β, AGE-RAGE–NF-κB, mitochondrial redox systems, and endothelial nitric oxide signaling. Precision-medicine and biomarker-guided models, such as the FINGER paradigm, illustrate emerging strategies that integrate metabolic and vascular profiling with neuroimaging and AI-driven analytics for patient-specific management.
Insulin resistance impairs cerebral glucose utilization, creating an “energy crisis” that facilitate amyloid accumulation and tau phosphorylation. Intranasal insulin therapy bypasses systemic metabolism to deliver insulin directly to the brain. A recent systematic review and Meta-Analysis demonstrated that intranasal insulin improved memory and attention in patients with mild cognitive impairment (MCI), with particularly strong effects in ApoE4 non-carriers (Long et al., 2022). Additionally, metformin an AMPK activator widely prescribed for type 2 diabetes has shown neuroprotective benefits. Observational studies report lower incidence of cognitive decline among metformin users, likely due to improved insulin sensitivity and reduced oxidative stress (Enderami et al., 2025). However, some data caution about possible vitamin B12 deficiency worsening cognition, emphasizing the need for balanced interpretation. Another promising frontier lies in GLP-1 receptor agonists (liraglutide, semaglutide), which enhance insulin sensitivity and exert anti-inflammatory effects in the CNS (Biessels & Whitmer, 2020). Preclinical studies revealed that liraglutide reduced amyloid deposition and improved synaptic plasticity in AD mouse models (Duarte et al., 2020). In a randomised clinical trial (N = 164531) shows GLP-1 receptor agonists was associated with significant reduction dementia (Seminer et al., 2025).
Furthermore, Pioglitazone is peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist used in type 2 diabetes management, has been investigated as a potential therapeutic agent in Alzheimer’s disease (AD) because of its insulin-sensitizing and anti-inflammatory effects (Alhowail et al., 2025). Activation of PPAR-γ improves glucose metabolism, reduces oxidative stress, suppresses pro-inflammatory cytokine production, and enhances amyloid-beta clearance. Experimental studies have shown that pioglitazone reduces neuroinflammation and improves cognitive performance in AD models (Blossom et al., 2026). However, clinical studies have reported inconsistent cognitive benefits, suggesting that therapeutic response may depend on disease stage and treatment duration.
Endothelial dysfunction restricts cerebral perfusion, leading to hypoxia, impaired glucose delivery, and reduced amyloid clearance. Restoration of nitric oxide (NO) signaling is central to reversing this pathology. Experimental studies have shown that supplementation with tetrahydrobiopterin (BH4) restores eNOS coupling, improves vascular tone, and enhances memory in AD mouse model (Fanet et al., 2021). Human evidence supports lifestyle-based endothelial protection, with aerobic exercise emerging as one of the most effective non-pharmacological strategies. Aerobic exercise trials reported increased hippocampal volume, improved perfusion, and higher brain-derived neurotrophic factor (BDNF) levels in older adults (Romero Garavito et al., 2025). These vascular benefits are accompany by neurobiological changes, including increased expression of brain-derived neurotrophic factor (BDNF), a key mediator of synaptic plasticity and neuronal survival. Furthermore, Statins which is prescribed for cholesterol lowering have been reported to exert additional vascular benefits by enhancing endothelial nitric oxide (NO) bioavailability and reducing oxidative stress (W. H. Chen et al., 2024). These pleiotropic effects help maintain cerebral perfusion and protect the blood–brain barrier. For example, a systematic review and meta-analysis indicated that long-term statin use is associated with a modest but significant reduction in dementia risk, particularly Alzheimer’s disease (Westphal Filho et al., 2025). Additionally, angiotensin receptor blockers (ARBs), particularly agents such as candesartan, extend beyond blood pressure control by exerting direct protective effects on cerebral vasculature. ARBs enhance cerebral perfusion and reduce vascular stiffness by inhibiting angiotensin II–mediated vasoconstriction, oxidative stress, and inflammatory signaling. These vascular improvements translate into better maintenance of blood–brain barrier (BBB) integrity and neuronal oxygen-glucose delivery (Zhou et al., 2023). Clinical evidence supports these benefits with small randomized trials and observational studies have reported that AD patients with hypertension treated with candesartan or related ARBs demonstrated slower rates of cognitive decline compared with those on non-ARB antihypertensives. ARB therapy was associated with improved scores on memory and executive function tests, highlighting the potential role of renin–angiotensin system modulation in neuroprotection (D’Silva et al., 2022).
Experimental evidence indicates that the receptor for advanced glycation end products (RAGE) serves as an important mediator connecting metabolic stress with neuroinflammation and amyloid accumulation. In animal models of Alzheimer’s disease, pharmacological inhibition of RAGE reduced microglial activation as depicted in Figure 4. The inhibition of oxidative stress, and decreased amyloid-beta deposition slow down the progression of neurodegeneration and offer neuroprotection (Derk et al., 2018). One of the most studied agents, azeliragon, a small-molecule RAGE inhibitor, progressed to phase III clinical trials. While the trials confirmed safety and tolerability, clinical outcomes revealed only modest improvements in cognitive performance compared with placebo, highlighting the limited efficacy of RAGE inhibition as a stand-alone therapy (Magna et al., 2023). These results suggest that RAGE antagonism may have disease-modifying potential. However, its clinical impact may be optimized in combination with other strategies, such as metabolic control in diabetes, lipid-lowering agents, or vascular protective drugs. This emphasised that targeting a single pathway is unlikely to be effective, and integrated strategies are needed to address the interconnected metabolic, vascular, and neurodegenerative mechanisms in Alzheimer’s disease (Taguchi & Fukami, 2023). In parallel, metabolic drugs such as GLP-1 agonists indirectly modulate AGE–RAGE signaling, lowering AGE-induced cytokine production. Lifestyle strategies, particularly low-sugar or Mediterranean-style diets, are associated with lower systemic AGE levels and reduced cognitive decline in longitudinal human cohorts. These data suggest that controlling inflammation through both pharmacological and non-pharmacological approaches could synergize with other therapies (Clark et al., 2022).
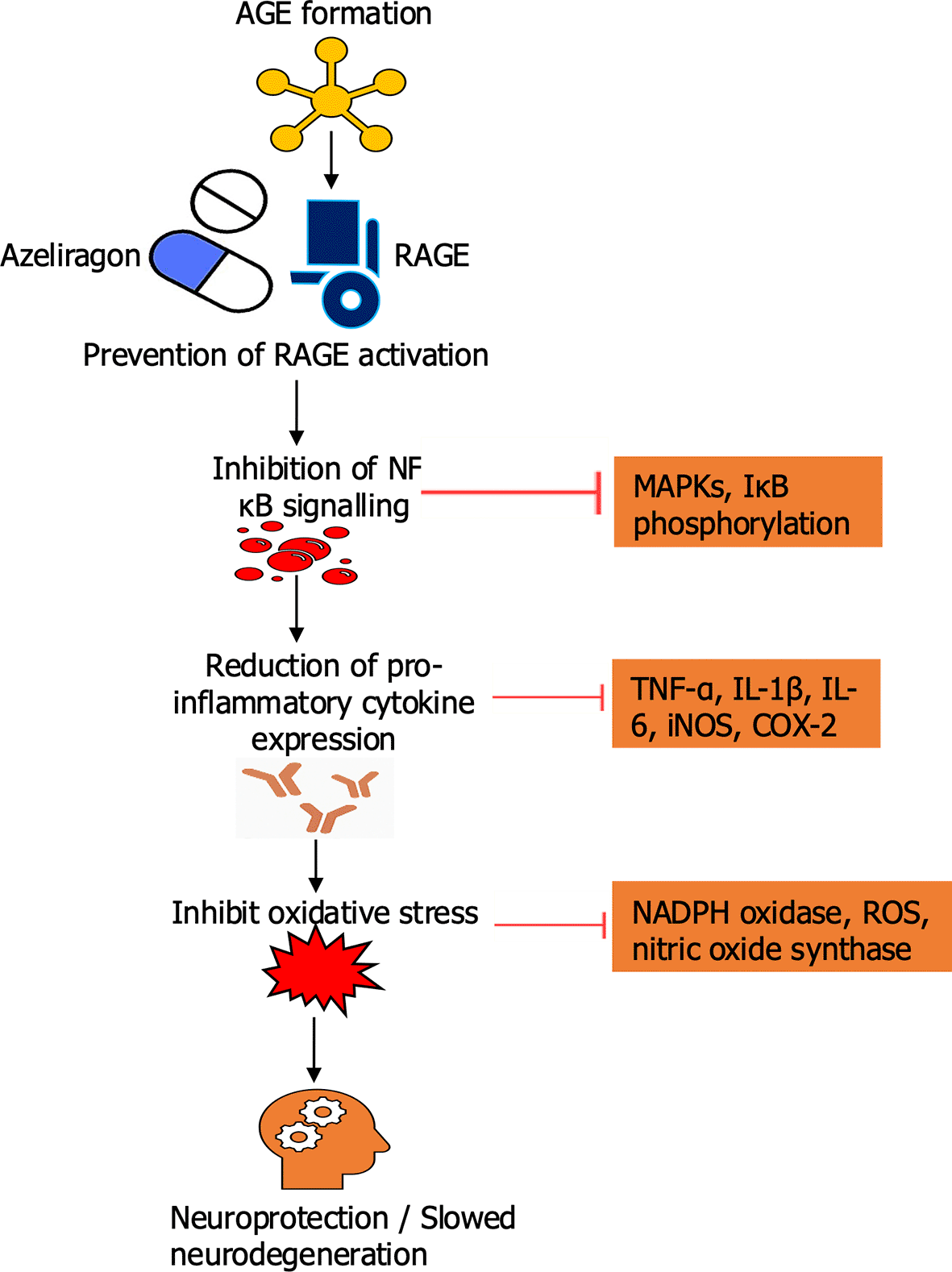
Schematic illustration showing the inhibitory effect of Azeliragon on the AGE–RAGE signaling pathway in Alzheimer’s disease. Under normal pathological conditions, binding of advanced glycation end-products (AGEs) to their receptor (RAGE) activates downstream NF-κB signaling, leading to cytokine release, oxidative stress, and subsequent amyloid-beta (Aβ) accumulation and neurodegeneration. Azeliragon blocks AGE binding to RAGE, preventing pathway activation and reducing neuroinflammation, oxidative damage, and Aβ-mediated neuronal injury.
Mitochondrial dysfunction is central to both metabolic disease and AD, producing ATP deficits and reactive oxygen species (ROS). Several experimental strategies aim to restore mitochondrial health. MitoQ, a mitochondria-targeted antioxidant, improved synaptic activity and reduced amyloid burden in mouse AD models (Zong et al., 2024). Similarly, the peptide SS-31 (elamipretide) preserved mitochondrial cristae structure, stabilized membrane potential, and enhanced memory in aged rodents. Human pilot trials of mitochondrial cofactors such as coenzyme Q10 and nicotinamide riboside have shown safety and modest cognitive improvement. Additionally, metabolic interventions like ketogenic diets also improve mitochondrial efficiency and reduce ROS generation, with preliminary human data showing improved cognition in mild cognitive impairment. These findings highlight mitochondria as both a therapeutic target and a metabolic regulator in AD (Zong et al., 2024).
The complexity of AD requires precision medicine guided by biomarkers. The Finnish Geriatric Intervention Study (FINGER trial) demonstrated that a multidomain lifestyle intervention diet, exercise, vascular risk control, and cognitive training slowed cognitive decline in elderly individuals at risk (Sakurai et al., 2025). Notably, biomarker sub-studies revealed improvements in metabolic and vascular markers, reinforcing the integrated approach. Modern biomarker platforms include CSF insulin, cholesterol, and AGE levels, alongside imaging modalities such as arterial spin labeling MRI to measure cerebral perfusion. When combined with amyloid and tau PET imaging, these tools stratify patients by metabolic and vascular risk, enabling therapies to be tailored. Additionally, artificial intelligence driven algorithms are being explored to integrate these diverse biomarker streams for real-time clinical decision-making (Sakurai et al., 2025).
Future research should emphasize the integration of network pharmacology and molecular docking to elucidate complex drug to target interactions across metabolic, endothelial, and neuroinflammatory pathways implicated in Alzheimer’s disease (AD). These computational approaches enable the identification of multi-target compounds capable of simultaneously modulating oxidative stress, mitochondrial dysfunction, and vascular injury. For example, in silico docking studies have revealed that certain phytochemicals and antidiabetic agents bind effectively to Aβ-aggregating enzymes and inflammatory receptors, suggesting their potential as dual-acting therapeutics (Prakash et al., 2023). Such integrated modeling accelerates drug discovery while improving the rational design of multi-pathway interventions.
Multi-omics technologies including genomics, transcriptomics, proteomics, metabolomics, and lipidomics present innovative means of dissecting the intricate molecular mechanisms linking metabolic dysfunction to neuronal degeneration. Recent omics-driven studies have identified plasma and cerebrospinal fluid (CSF) biomarkers (Cardillo et al., 2025). These include lipid peroxidation products and inflammatory metabolites, that correlate with early cognitive impairment. Integrating these omic layers enables the development of composite biomarker panels for early diagnosis, prognosis, and therapeutic monitoring. This approach will help clinicians stratify patients based on molecular phenotypes, leading to more precise and personalized interventions in AD management (Liu et al., 2025).
Artificial intelligence (AI) and machine learning hold transformative potential for unraveling the complexity of Alzheimer’s disease. AI algorithms can detect subtle, preclinical patterns of neurodegeneration before clinical symptoms emerge. Predictive models developed through deep learning frameworks have already demonstrated high accuracy in forecasting disease progression and therapeutic responsiveness. Future AI-driven systems could guide real-time clinical decision-making, enabling dynamic, personalized treatment adjustments and enhancing the efficiency of clinical trials (Kale et al., 2024).
Drug repurposing represents a cost-effective and time-efficient strategy for developing new treatments targeting metabolic and endothelial pathways in AD. Compounds such as metformin, pioglitazone, and statins have shown promising neuroprotective and vasoprotective effects beyond their primary indications. Network pharmacology analyses reveal that these agents act on shared molecular hubs including AMPK, NF-κB, and eNOS signaling emphasizing the value of polypharmacology in addressing multifactorial disease mechanisms (T. Chen et al., 2023). Future therapeutic strategies should therefore focus on combination or multi-target drugs that can simultaneously modulate metabolic stress, vascular dysfunction, and neuroinflammation.
Bridging the gap between molecular discoveries and clinical application remains an Important challenge. Translational frameworks that combine computational modeling, omics-based diagnostics, and controlled clinical trials are needed to validate mechanistic hypotheses and therapeutic efficacy. Collaborative consortia linking academic institutions, pharmaceutical industries, and bioinformatics platforms will facilitate large-scale data integration and reproducibility. Ultimately, these efforts will promote a shift toward precision neurotherapeutics where treatment is tailored to each patient’s genetic, metabolic, and vascular profile improving outcomes in Alzheimer’s and related neurodegenerative disorders.
Alzheimer’s disease (AD) is recognized as a systemic disorder in which metabolic dysfunction and endothelial injury act synergistically to amplifier neurodegeneration. Insulin resistance, dyslipidemia, and hyperglycemia impair neuronal energy metabolism, while endothelial dysfunction and blood–brain barrier breakdown amplify oxidative stress, inflammation, and amyloid pathology. This interconnection defines a metabolic–vascular–neurodegenerative axis that links systemic disease to brain pathology. Effective management therefore requires a paradigm shift from single-target therapies toward integrative, multi-system interventions that restore metabolic balance, protect vascular integrity, and mitigate neuroinflammation. Emerging strategies from insulin sensitizers and endothelial stabilizers to mitochondria-targeted agents offer promising translational opportunities for disease modification. Addressing Alzheimer’s disease through combined metabolic and vascular interventions such as improving insulin sensitivity, reducing oxidative stress, and preserving endothelial integrity offers a more effective, system-level strategy for preventing cognitive decline and promoting long-term brain health.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)