Keywords
Creutzfeldt-Jakob syndrome; Prion diseases; differential diagnosis; behavioral symptoms; 14-3-3 Proteins; Peru
Creutzfeldt-Jakob syndrome; Prion diseases; differential diagnosis; behavioral symptoms; 14-3-3 Proteins; Peru
The revised version of the manuscript incorporates the reviewers’ suggestions. The Introduction has been updated to include the most recent literature related to the topic. The MRI image has been replaced with an improved version to enhance clarity and understanding of the content. Additional relevant details have been added to the case description. Minor modifications have been made to the references, and the text has undergone a grammar review to improve coherence and readability.
See the authors' detailed response to the review by Anna Ladogana
See the authors' detailed response to the review by Shi-Lin Yang
Creutzfeldt-Jakob disease (CJD) is one of the main prion diseases, an always fatal neurodegenerative disorder.1 The common pathological process is characterized by the conversion of the normal cellular prion protein (PrPc) to an insoluble anomalous form (PrPSc) that accumulates progressively in the brain, sometimes forming extracellular amyloid plaques.2
CJD is classified, according to the etiology, into sporadic (idiopathic or classic) (85%), acquired (or exogenous) (5%), genetic (or hereditary) (10%), and other variants such as Gerstmann-Sträussler-Scheinker disease (GSSD), fatal familial insomnia (FFI) and kuru.1,3,4
The probable diagnosis is based on progressive dementia and other clinical criteria (myoclonus, visual disorders, cerebellar signs, pyramidal or extrapyramidal signs, and akinetic mutism), as well as a compatible electroencephalogram (EEG) (periodic epileptiform discharges) and/or detection of neuronal protein related to neuronal destructions that accumulates in cerebrospinal fluid (CSF), called protein 14-3-3.1,3,5 The definitive diagnosis requires a post-mortem study.6
Due to these clinical complexities and the variable initial onset, it presents a common delayed detection with issues during the differential diagnosis. In this paper, we include a representative case from our institution and discuss the common challenges during the course of the disease.
We present a case report from the “Hospital Regional Lambayeque”, in Lambayeque, Peru. This report followed the CARE guidelines.7
We present the case of a 47-year-old male from the Amazon region of Peru, unemployed, with no pathological or surgical history. His symptoms start with oppressive non-located headache 80-weeks before, which were treated with analgesic medication without response. It was associated with a hands rest tremor, perceptual alteration of time, difficulty in recognizing relatives, aggressiveness, agitation, and mild memory impairment.
24-weeks before, symptomatology persists and progress until presenting alterations of sphincter control and difficulty for walking, and upper limb rest and intention tremor generating partial dependence for their daily activities. On detailed neurological examination, the patient presented both extrapyramidal (rest and intention tremor, and bradykinesia) and pyramidal signs (generalized hyperreflexia and bilateral Babinski sign). Gait instability with intention tremor suggested cerebellar involvement, and intermittent involuntary jerks observed during hospitalization were compatible with early myoclonus. He had a hospital admission for presenting focal to bilateral tonic-clonic seizures for which he received antiepileptic drugs with an incomplete response. Subsequently, due to the persistence of symptomatology and the development of epileptic status, he was hospitalized again. The blood studies showed anemia (10.2 g/dl), with any other abnormalities. The CSF analysis found a clear fluid without alterations.
Brain magnetic resonance imaging (MRI) showed moderate to severe cortico-subcortical atrophic changes and subcortical hyperintensities ( Figure 1). Additionally, an EEG was performed, without any alteration. Due to the progressive neurological compromise and the combination neurological findings, patient met the requirement of at least two supportive signs (pyramidal/extrapyramidal and cerebellar/myoclonic features) as defined in the diagnostic criteria for probable CJD. Moreover, the development of early and progressive dementia in the next months, and the MRI suggestive of prion disease, CJD diagnostic was prioritized for exploration. CSF 14-3-3 protein was analyzed by a semi-quantitative Western blot assay in an external reference laboratory. Patient CSF was run alongside negative and positive control samples, and band intensity for 14-3-3 was densitometrically compared to these controls. According to the laboratory’s validated cut-off, the result was classified as strongly positive (‘high titre’), supporting the diagnosis of probable CJD. Symptomatic treatment and palliative care were indicated to the patient.
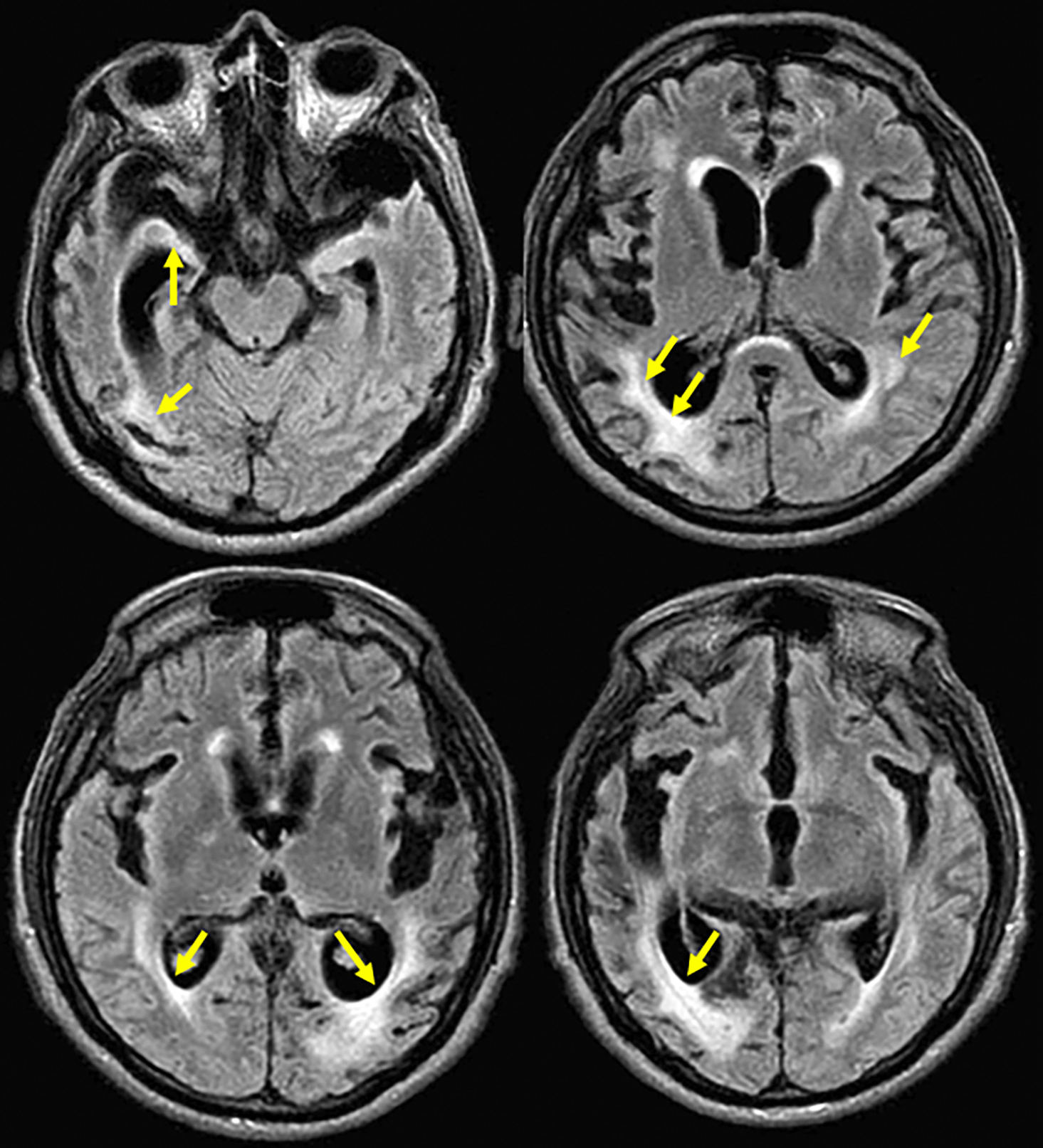
Axial section in a fluid attenuated inversion recovery (FLAIR) sequence showing hippocampal volume decreased in both temporal lobes with increase of temporal horns predominantly on right side; mild hyperintensity in right caudate nucleus and moderate in periventricular regions with confluent zones in the right parietal region; and moderate to severe atrophic cortical-subcortical with frontotemporal predominance (Hyperintensities highlighted with arrows).
All these tests were performed during the hospitalization of the patient. The Western blot test of the protein 14-3-3 was performed two weeks after his hospitalization. The patient was evaluated one month after discharge, but he did not return for his next consultation, and it was not possible to follow up on his case.
The case reported a progressive neurological condition (headache that progress to dementia) with early onset of cognitive impairment and behavioral symptoms at the age of 47 years, a Brain-MRI showing moderate to severe cortico-subcortical trophic changes, and basal nuclei hyperintensity, added to a positive Western-Blot quantification of CSF 14-3-3 protein (sensitivity of 94% and specificity of 84%),5,8 hence the condition was classified as spongiform encephalopathy, probable CJD. It was not possible to make the definitive diagnosis with brain biopsy confirmation, where neuronal loss, gliosis, and intracytoplasmic vacuolization are observed in the brain parenchyma.6,9
CJD is a rare and fatal neurodegenerative disease, due to the high mortality, an early and timely diagnosis must be made.10,11 However, currently the diagnostic criteria are controversial and there are several variances of the onset symptoms, thus, the diagnosis is usually made in the terminal phases of the disease.
We search the available literature to compare with our experience and found ten articles reporting diagnostic challenges in CJD ( Table 1).12–21 The age range of the patients reported was 30 to 86 years [median 64 years]. The main initial clinical manifestation was psycho-behavioral symptoms (50% of the cases), predominantly: apathy, confusion, aggressiveness, irritability, and sleep disturbance. The initial differential diagnoses were psychiatry exacerbation (depression and schizophrenia), myelopathy, epilepsy, stroke and parkinsonism. Our patient is 47-years old, male, with an initial presentation of headache, extrapyramidal signs, and behavioral symptoms. In another case series,22 they found that the presentation was nonspecific, from headache to ataxias. The extrapyramidal and cerebellar signs onset is presented as a frequent initial manifestation.23 These data coincide with our systematic review results, indicating a common non-specific initial symptomatology in these cases.
The initial neuroimaging evaluations were reported in nine of the cases,12–14,16–21 mainly, they found hyperintensity in frontotemporal lobes and cortico-subcortical atrophy. EEG evaluations reporting theta range with slower delta rhythms.
The broad clinical findings spectrum included a progressive neuropsychiatric syndrome –whose initial symptoms could be depression, insomnia, anxiety, apathy, and hallucinations. Besides, movement disorders– cerebellar ataxia, involuntary movements, myoclonus, chorea, and dystonia- and persistent pain or paresthesia, could be associated. Finally, dementia and akinetic mutism could be presented at the end of this disease.
In general, psychiatric symptoms precede neurological manifestations.9,22,24 We found that 65% of cases start clinical manifestation with psychobehavioral symptoms; it is according to our case report.
Timely diagnosis of CJD is often difficult, in part because of the frequency of unusual variants, with potential modifications due to early interventions. The available literature on this topic shows a wide diagnosis delay range from four to 80 weeks [median 14 (5-72 weeks)]. The best approach is to add CJD as our differential diagnosis options, not only in the classical rapid-progressive dementia diagnosis, but also in front of patients with suspicious of psychiatry exacerbation, myelopathy, epilepsy, stroke, and Parkinsonism.
The reports show that autoimmune encephalitis due to Hashimoto’s thyroiditis and a CJD-like syndrome due to parathyroid adenoma could simulate the CJD clinical manifestations even meeting partially the diagnostic criteria.25 Thus, it is important to evaluate carefully the autoimmune disease history in patients with CJD suspicious.
Current reviews indicate that the changes in brain-MRI are bilateral, symmetric, and predominant in basal ganglia and cortical regions,6,26 in addition, 85% show signs of atrophy in the MRI.6 Our case findings are according to this literature, however, in most of the cases with difficult timely diagnosis, the MRI results were inconclusive or normal until the end-stage of the disease, and the most common finding was cortical-subcortical atrophy and basal nuclei hyperintensity.
The patient presented unspecific characteristics of onset, in addition to a delay of 80-weeks before CJD diagnosis, with broad clinical manifestations, alterations in the MRI and the presence of protein 14-3-3 in CSF.
Although the patient had no reported family history of neurodegenerative or prion-related disorders, PRNP gene sequencing was not performed due to limited availability. Therefore, while the clinical and paraclinical findings strongly supported sporadic CJD, a hereditary prion disease cannot be definitively excluded.
In conclusion, not all the CJD symptoms are at the beginning of the disease, and not all the classical symptoms and signs indicate CJD. In addition, as seen in the review, the delay to a proper diagnosis of this disease is large, so this is a significant diagnostic challenge for physicians and neurologists. CJD should be included as a differential diagnosis at the beginning of behavioral symptoms or rapid cognitive impairment, even if it is a remote possibility.
Conceptualization: VEFR, YKYB, GAM, CAD, KPB; Data curation: VEFR, KPB; Investigation: VEFR, YKYB, GAM; Methodology: VEFR, ANF, CAD, KPB; Supervision: KPB; Writing – Original Draft Preparation: All authors; Writing – Review & Editing: All authors.
This study was conducted according to the ethical standards of the Declaration of Helsinki. Informed consent was obtained from the patient and this case study was approved by the ethical board of Hospital de Lambayeque. Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)