Keywords
Biomarkers, algorithmic, AI, evolutionary computation, precision medicine, clinical trial, anti-TNF, rheumatoid arthritis
This article is included in the AI in Medicine and Healthcare collection.
Biomarkers, algorithmic, AI, evolutionary computation, precision medicine, clinical trial, anti-TNF, rheumatoid arthritis
We appreciate the opportunity to revise our paper. We have carefully reviewed and commented on the reviewer comments. We have made minor revisions to address specific points raised by the reviewers. Some reviewer comments were quite general and did not provide enough specificity to directly address. An important point, is that we present an example of an algorithm to allow independent verification of our approach. To our knowledge, such transparency is very unusual, if not unique, in publications of this nature. Hence, we consider that to be a particular strength of the publication that will raise the bar for the future.
See the authors' detailed response to the review by Daniel H. Solomon and Kathryne Marks
See the authors' detailed response to the review by Jean Roudier
The completion of the Human Genome Project in 2003 was accompanied by optimism that a profusion of novel diagnostics and therapeutics would rapidly ensue. Technical innovations and digitization have since produced vast amounts of data without clearly impacting the trajectory of clinical advances.1 The lack of predictive biomarkers with sufficient accuracy to inform therapeutic decisions for individual patients is an unresolved problem, thwarting the realization of the vision of precision medicine.2,3 Why has the explosive growth in biomedical data not enabled greater progress, especially with respect to the discovery and development of predictive biomarkers? The typical explanation is the daunting complexity of biology.
Living organisms are complex systems, containing orders of magnitude more information than non-living entities.4 Biological functions, including disease and drug responsiveness, result from complex networks of cellular and molecular interactions that can potentially be described mathematically, reflecting mechanisms and constraints under which biological systems operate.5–9 These interactions are typically nonlinear and multi-dimensional which complicates their analysis and definition.10–13 This explains why single variable biomarkers are typically inadequate providers of clinically useful predictive insight.11
Identifying causal factors from data is an inverse problem. What would an ideal analytic solution to the inverse problem as applied to biological complexity to yield actionable insight, including predictive biomarkers, look like? In a complex system, information is present in the relationships between components, as well as in the individual components themselves. An effective analytic solution would reveal the specific molecular networks that underly biological functions, diseases and therapeutic effects, respectively. The analytic approach would not assess the components in isolation. Instead, it would identify the relevant components in terms of their mathematical relationships with other components to reflect accurately their frequently non-linear nature. In essence, the solution would distil essential information into transparent explanatory and predictive quantitative algorithms, which could then be translated into clinical tests. Such an analytic solution could be applied to all forms of biomedical data, including clinical trial data. Randomized clinical trial outcomes are conventionally analyzed using a pre-specified hypothesis and statistical tests, based on the average overall response, in order to assess whether treatment is effective at a population level. Much information from clinical trials is neglected with this approach, resulting in a lack of guidance for clinicians and patients as to which individuals might benefit from the therapy. The ability to summarize clinical trial data in terms of transparent summary, quantitative, easily validated algorithms predictive of individual patient outcomes would have significant potential for informing both clinical practice and research.
Since machine learning is not adequate, in part because of its black box solutions, and in part because prevalent methods do not have a broad palette of mathematical functions to characterize many interactions present in biological systems, we developed a novel approach.14,15 We devised a novel analytic solution based on an evolutionary computation foundation fused with mathematics, the science of complex emergent systems, information theory and its subset, algorithmic compression theory. The software incorporated a comprehensive set of mathematical functions, to produce transparent interpretable predictive algorithms from complex biomedical data, specifically designed to model the nonlinear and high dimension relationships that define complex emergent systems. The algorithms produced are verifiably accurate mathematical solutions that can predict the outcome of interest.
To test the software, we applied it to a previously published placebo-controlled clinical trial of infliximab in RA, conducted by a pharmaceutical company with baseline peripheral blood transcriptomic data.16,17 Infliximab is a monoclonal anti-TNF antibody effective in treating a range of immune mediated disease including RA. The hypothesis was that the software would produce transparent, interpretable, discovery algorithms predictive of individual treatment responses that could be independently validated. We selected this trial because it was randomized and placebo controlled, with tens of thousands of RNA data points available for each patient to provide a rigorous test. The software produced a discovery algorithm comprised of biomarker measurements, a clinical variable in addition to mathematical functions, with 100% accuracy in predicting both infliximab and placebo treatment outcomes. The biomarkers were four RNA gene expression variables. The discovery algorithm is shown, in different formats, to enable independent validation. When those four gene expression variables in the original algorithm were excluded from subsequent analyses, using exactly the same analytic approach, additional algorithms were derived, with four different gene expression variables and similar 100% predictive accuracy.
Having identified the eight discovery gene expression variables, we then hypothesized that application of the software, using only those eight variables to previously published data from six additional RA studies containing baseline gene expression data, would also derive accurate predictors of clinical outcomes following anti-TNF treatment. In each case, using just these eight gene expression variables, the software provided an algorithm more predictive of treatment response than reported in the original publications, thereby independently validating both the gene expression variables as response predictors, and the superior predictivity of our methodology relative to the various approaches, including different variants of machine learning, previously reported. The gene expression variables we discovered are now in development as a clinical test, which will need to be validated prospectively in future studies for clinical use.
The discovery cohort was from a published randomized, double-blind placebo-controlled trial of infliximab, in RA patients naïve to biologics therapy following an inadequate response to methotrexate.16,17 Participants were recruited between April 6th, 2011 and March 29th, 2012 from three European clinical centers: one in Romania and two in Moldova.17 The study was conducted in accordance with principles of Good Clinical Practice and was approved by the National Ethics Committee in Romania and the National Ethics Committee, Clinical Research of Drugs and Methods of Treatment in Moldova, with all subjects providing informed written consent.17
Active disease was defined as at least 6 tender and 6 swollen joints, with a rheumatoid arthritis magnetic resonance imaging score of ≥1 in the radio-carpal or intercarpal joints, as objective confirmation of disease activity. The participants were all on stable doses of methotrexate, steroids, and/or non-steroidal anti-inflammatory drugs. At weeks 0, 2, 6, and 14, the participants received either infliximab 3 mg/kg or placebo. The participants were of mean age of 50 years, predominantly female (92%), and rheumatoid factor positive (91.5%), with a mean baseline Disease Activity Score-28 (DAS28) score of 6.2. The primary endpoint was a magnetic resonance imaging assessment of disease activity. This endpoint was not used in our analysis, because the imaging data were not available for individual participants. The study had 80% power to yield a significant difference in DAS28 score.17 The trial used the European League against Rheumatism (EULAR) DAS28 score to evaluate response, defined as a decrease of 1.2, as a binary dependent variable for the analysis at 14 weeks, yes or no. Baseline peripheral blood gene expression data for 59 patients were available plus one additional variable reflecting treatment, either infliximab or placebo, resulting in a total of 52,379 potentially independent variables.16 The participant flow is shown diagrammatically in the original publication, where the study protocol is referenced.17
To independently validate the gene expression variables in the discovery algorithms, we applied these variables to 6 previously published studies with available baseline gene expression data from patients with active RA, treated with anti-TNF therapies.18–23 These studies were done in different geographies, used different methodologies to process the samples and gene expression data, and in some cases, used different anti-TNF therapies but with a similar 14 week efficacy primary endpoint, except for one study.18–23 Summary details on the discovery and validation studies are provided in Table 1, with details available in the original publications.18–23 Written informed consent for all participants and local ethics approval was obtained for each of the studies that provided validation data as detailed in the original publications.18–23
| First author | MacIsaac et al. (2014)16,17 | Lequerre et al. (2005)18 | Julia et al. (2009)19 | Bienkowska et al. (2009)20 | Toonen et al. (2011)21 | Nakamura et al. (2016)22 | Tanino et al. (2010)23 |
|---|---|---|---|---|---|---|---|
| GEO accession | GSE58795 | GSE3592 | GSE12051 | GSE15258 | GSE33377 | GSE78068 | GSE20690 |
| Phase | Discovery | Validation | Validation | Validation | Validation | Validation | Validation |
| Location | Moldova & Romania | France | Spain | USA | Netherlands | Japan | Japan |
| RA classification criteria | RA ACR (1987) | RA ACR (1987) | RA ACR (1987) | RA (1987) | RA ACR (1987) | RA ACR (1987) or EULAR/ACR (2010) | Not reported |
| Treatment | infliximab or placebo | infliximab | infliximab | infliximab adalimumab, and etanercept | infliximab and adalimumab | infliximab | infliximab |
| RA treatment population | MTX resistant. No prior TNF therapy | MTX resistant. DAS28>=5.1 | MTX resistant. DAS28>3.2. No prior TNF therapy | Active RA: No TNF in past 6 months | DMARD resistant. DAS28>3.2. No prior TNF therapy | MTX resistant | MTX resistant |
| Platform | Rosetta/Merck custom Affymetrix 2.0 microarray | Affymetrix Human Genome U133A Array | Illumina H-6 mRNA Sentrix Human-6 Expression BeadChip | Affymetrix Human Genome U133 Plus 2.0 Array | Affymetrix GeneChip Exon 1.0 ST | Agilent-014850 Whole Human Genome Microarray 4x44K G4112F | Agilent-014850 Whole Human Genome Microarray 4x44K G4112F |
| Efficacy | 14-week EULAR criteria | 14-week EULAR criteria | 14-week EULAR criteria | 14-week EULAR criteria | 14-week EULAR Criteria | 6-month CDAI | 14-week serum CRP |
The gene expression profiles analyzed were downloaded from the GEO database. Because all the data used were previously published, de-identified and publicly available, neither ethics committee approval nor informed consent were required. Datasets were downloaded and transposed so that gene expression values and clinical variables were changed from rows to columns, and subject records were changed from columns to rows. Data files were saved in CSV format and imported into the software for analysis, without any pre-processing of the biomarker measures.
The software is a quantitative analytic platform based on evolutionary computation, designed as a scalable, unbiased methodology to produce transparent algorithms based on mathematical relationships from complex data, without any prior assumptions other than the patient selection criteria and designs of the original studies yielding data for analysis. The software fuses evolutionary principles, signal processing functions, and information theory, and requires no domain expertise or prior knowledge of the nature of a problem in terms of explanatory variables, dimensionality or underlying mathematical relationships. A distinctive feature is that the software uses all available data to derive the algorithms, without any filtering process to exclude variables based on commonly used thresholds or feature selection methods. This enables the identification of variables typically discarded by feature selection methods used in biomarker discovery. This includes such methods’ tendency to discard potential explanatory variables, with relatively low expression or nonlinear relationships to the outcome, but which may be functionally important because of the nonlinear and binary threshold interactions pervasive in complex biologic systems. The software identifies key variables in the context of their mathematical relationships with each other and associated with outcomes of interest, and is not dependent on baseline differences in biomarker levels between responders and non-responders. The software automatically excludes overfitted algorithms and automatically divides the data into three distinct, random subsets that are sequentially processed: a training set, a selection set, and a test set. Analysis of the training subset provides an ensemble of candidate algorithms, which are then evaluated on the selection subset, to select a final algorithm, which is then validated on the test set. An overfitted algorithm would not be validated on the test set, ensuring that overfitted algorithms are not selected. The training, selection, and test data subsets are scrupulously segregated, to avoid any information leakage between the discrete components of the process. In the discovery analysis of the MacIsaac et al. data, there were 18 patients in the training set, 21 in the selection set and 20 in the test set ( Table 2).
| Analysis of data from MacIsaac et al.16 | |
|---|---|
| Training set: 9 placebo, 9 infliximab | |
| Errors/total | 0/18 |
| Accuracy | 100% |
| Misclassification rate | 0% |
| Validation set: 11 placebo, 10 infliximab | |
| Errors/total | 0/21 |
| Accuracy | 100% |
| Misclassification rate | 0% |
| Test set: 9 placebo, 11 infliximab | |
| Errors/total | 0/20 |
| Accuracy | 100% |
| Misclassification | 0% |
| Overall Accuracy | 100% |
| Overall Sensitivity | 100% |
| Overall Specificity | 100% |
| Number of gene expression variables | 4 |
The 8 variables in the discovery algorithms were then applied to the analyses of six additional published data sets in patients with RA, using both baseline gene expression data and response outcomes to anti-TNF therapy, for independent validation. The intent was to prospectively validate the 8 variables and also to benchmark the predictivity of the algorithmic biomarkers that incorporated them, relative to prior predictors using traditional analytic approaches, including machine learning.
The software initially yielded an algorithmic biomarker set with five variables and twelve sequential mathematical instructions as shown in Table 3. There were four gene expression variables expressed quantitatively: SPTY2D1, Clorf105, KCTD4 and UL84 and the fifth variable was treatment assignment: infliximab or placebo. The algorithm is also presented in the form of an equation in Figure 1a, and in schematic form in Figure 1b. The three different depictions of the algorithm allow transparent validation of the discovery algorithm using the MacIsaac et al. GSE58795 dataset.
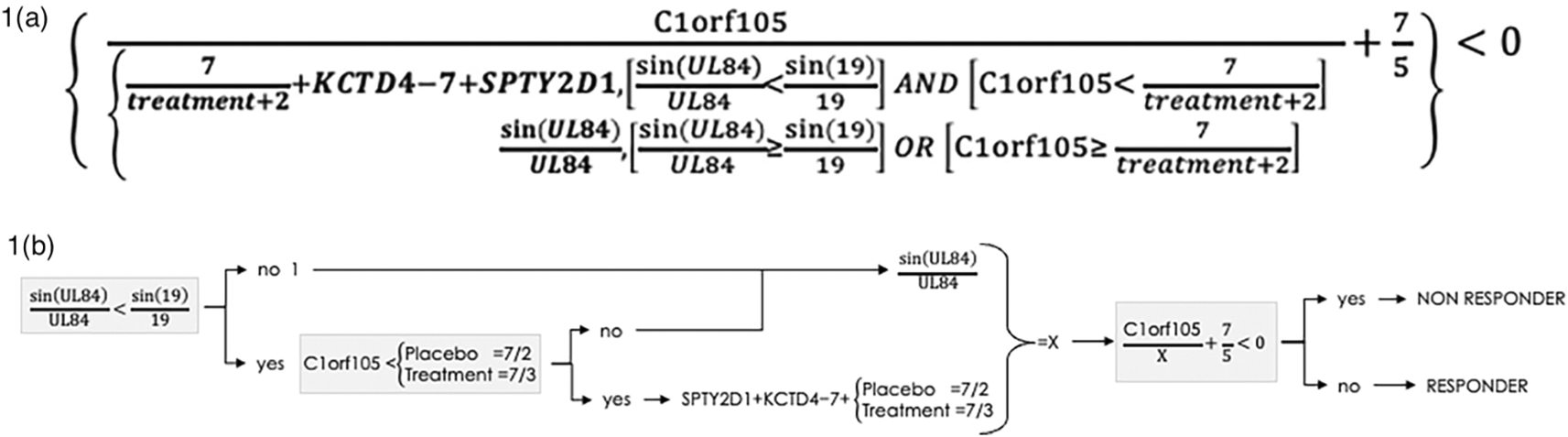
The discovery algorithm containing 4 variables is transparently depicted as an equation and also in schematic form to complement its depiction as a sequence of operations in Table 3, to facilitate both understanding and independent validation using the data from GSE58795 from MacIsaac et al. accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58795.16
A calculation for an individual patient, with a resulting value of less than zero indicated treatment non-response, and a value of zero or more indicated treatment response. The algorithmic variables were not those with the highest levels of expression. Agnostic evolutionary selection of treatment as a variable, either infliximab or placebo, into the predictive algorithm is evidence that the treatment has a mathematically significant impact on the response outcome for some patients. Although expected a priori, the treatment assignment variable was selected agnostically by the evolutionary process, and not pre-specified. Users of the software cannot influence or require any variable to be incorporated into an algorithm.
The performance metrics for the components of the discovery analyses are shown in Table 2. The overall accuracy was 100%, with 100% sensitivity and specificity. Repeat analyses consistently yielded exactly the same variables, and the mathematical instructions encoded in all algorithms were, without exception, mathematically equivalent in terms of binary outcome: response or non-response. Omission of any of the mathematical instructions from the algorithm degraded predictivity. Therefore, the algorithm was optimized and devoid of superfluous calculations. Accuracy and reliability were consistent across training, validation and test sets analysed. When the initial four gene expression variables were excluded from subsequent analyses, four additional gene expression variables were identified as components of algorithms, also with 100% accuracy: PTPRC, TPM3, ARHGDIB and SIAH1 ( Table 4). The software selects the minimum number of variables for maximum accuracy. The fact that a second group of four variables provided algorithms with 100% accuracy implies that these variables are highly correlated with the original four, containing almost as much information.
| Expression Variable | Function | |
|---|---|---|
| SPTY2D1 | Suppressor of Ty 5 homolog | Tumor suppressor gene in thyroid cancer24 |
| C1orf105 | Chromosome 1 open reading frame | Vascular remodeling, coronary artery disease and atrial fibrillation25,26 |
| KCTD4 | Potassium channel tetramerization domain containing 4 | Expressed in sepsis and esophageal cancer27,28 |
| UL84 | Gene for human CMV protein UL84 | Prior CMV infection associated with treatment response in RA29,30 |
| PTPRC | Protein Tyrosine Phosphatase Receptor Type C or CD45 | Genetic variants associated with anti-TNF response in RA31,32 |
| TPM3 | Tropomyosin 3 | Tropomyosin isoform associated with neoplasia, myopathy and as an autoantigen in RA33–35 |
| ARHGDIB | Rho GDP Dissociation Inhibitor Beta | Negative regulator of Rho guanosine triphosphate (RhoGTP) ases, reduced in osteoarthritis synovial fluid and elevated in early RA synovial fluid36,37 |
| SIAH1 | Siah E3 Ubiquitin Protein Ligase 1 | Downregulated in both RA and tuberculosis, may influence amplitude of inflammatory gene response38 |
We then tested the hypothesis that the eight variables from the discovery algorithms could be used to derive algorithms predicting individual treatment anti-TNF response outcomes from six additional published RA clinical trial data sets, providing independent validation of the variables.18–23 These data sets were chosen because they were derived from clinical trials similar to the discovery trial, though none were placebo controlled. While the eight variables used were the same for all six dataset analyses, distinct algorithmic operations were necessary for each validation, because of variation in quantitation instruments, measurement scale, data preparation and normalization methods used by the original researchers across the individual datasets. Summary information on the clinical studies is shown in Table 1, with further details available in the original publications.18–23 In each case, the algorithms we produced had superior performance to the analyses presented in the original publications as shown in Table 5, even though only a subset of the eight discovery variables were available in some datasets. The original analyses used a wide range of analytic methods including the most common machine learning approaches: neural networks, support vector machines and random forests.
| First author | MacIsaac16 | Lequerre18 | Julia19 | Bienkowska20 | Toonen21 | Nakamura22 | Tanino23 |
|---|---|---|---|---|---|---|---|
| Number of patients | 59 | 30 | 44 | 46 – leaving out intermediate responses or 75 including intermediate responses | 42 | 140 (infliximab treated) | 68 |
| Classifier Method | OLS regression | UHC | SVM, DDA, RF, & k-NN | RF | K-means cluster analysis | Logistic regression | LOO |
| Cross-validation analyses | None | LOO | LOO | LOO, NN, LDA and SVM | None | None | None |
| Original reported results | None reported | Sens 90% Spec 70% | Sens 94% Spec 86% | Sens 88% Spec 84% | Sens 61% Spec 71% | Sens 79% Spec 47% | Sens 68% Spec 86% |
| Current analysis | Sens 100% Spec 100% | Sens 100% Spec 100% | Sens 100% Spec 86% | Sens 100% Spec 86% | Sens 100% Spec 96% | Sens 95% Spec 91% | Sens 96% Spec 84% |
The weighted-average sensitivity and specificity of the individual models from the analyses we conducted across all 7 datasets, with a total of 400 subjects treated with anti-TNF therapy, were 98.5% and 90.9% respectively, surpassing the accuracy of all the prior published individual analyses, as shown in Figure 2. Accuracy for the marketed TNF response predictor from two reports, Mellors et al. and Jones et al., is also presented for comparison.39,40 The marketed predictor consists of 23 variables and was derived using machine learning, with a combination of neural networks and random forests. The actual model has not been published.
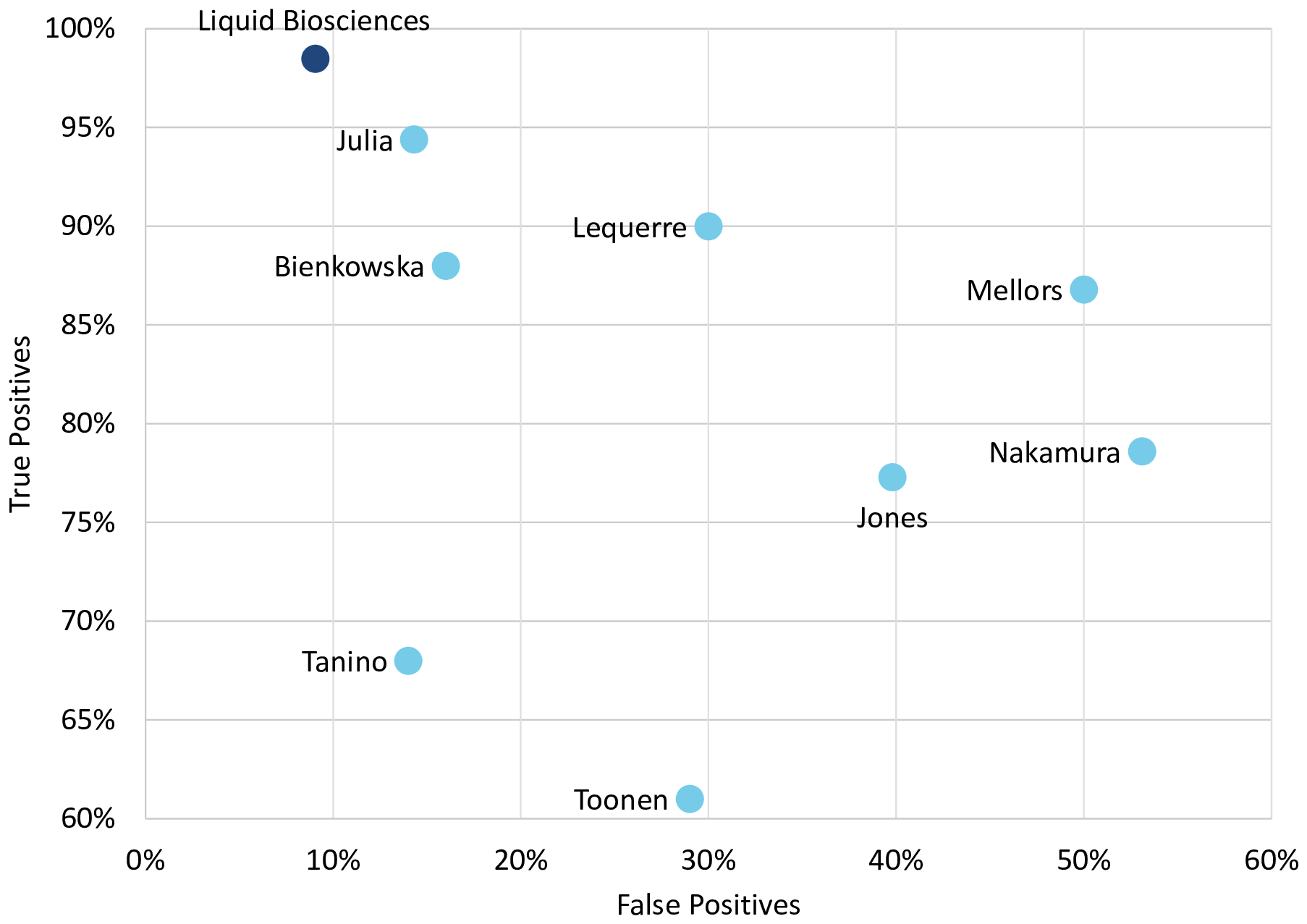
Weighted-average accuracy of the individual analyses we conducted using 7 datasets, on all 400 subjects receiving anti-TNF therapy had 98.5% sensitivity and 90.9% specificity as shown. This surpassed the performance metrics for all the individual previously reported analyses which are also shown.18–23 No metrics were provided for MacIsaacs et al. study used for discovery.16,17 Accuracy for a marketed TNF predictor from two reports, Jones et al. and Mellors et al., is also presented for comparison.30,31
To address the stagnation in biomarker discovery and development we applied a novel analytic approach, based on evolutionary computation and information theory incorporating mathematical functions, to a placebo controlled randomized clinical trial with 52,379 baseline gene expression variables for each of 59 patients. This provided discovery biomarker algorithms perfectly predictive of individual responses to both active therapy and placebo. The algorithms contained subsets of 4 variables from a total of 8 variables. The eight discovery variables were then validated, using the same analytic approach, as components of algorithmic predictors when applied to data from six independent clinical trials with different TNF inhibitors, on 3 continents, in multiple ethnicities, despite differences in response criteria, gene expression processing platforms and measurement scale differences, thereby providing extremely high confidence as to their validity.
The MacIsaacs et al. clinical trial study that provided the discovery data, was conducted by a major pharmaceutical company but did not provide a predictor of therapeutic response, nor did it identify any of the 8 variables we discovered as predictive.16 In each of the six validation analyses, our derived algorithms all containing 4 or fewer variables, had sensitivity and specificity superior to benchmarks in the original publications, which used a variety of mainstream analytic approaches including several types of machine learning.
None of the publications for the six validation datasets identified the eight predictive algorithmic biomarkers we discovered18–23 or reported an algorithm that might be the basis for a predictive test. To enable conclusive independent confirmation of our novel approach, we show an example of a perfectly predictive discovery algorithm, depicted three different ways: as a series of computer instructions, as an algebraic equation and also in schematic form.
The performance metrics for the algorithms we report surpass those of the marketed predictor of anti-TNF response derived using machine learning, consisting of 23 variables, which only identify non-responders with a specificity of 77.3-86.8% and sensitivity of 50.0-60.2%.39,40
Precision medicine requires highly predictive biomarkers to inform treatment decisions for individual patients. With the availability of more therapeutic options, selecting the most appropriate for an individual patient is an increasingly difficult challenge. Clinicians currently rely on trial and error approaches.
The necessary biomarkers, to align patients with the optimal therapy, have not been forthcoming despite huge increases in the production of biomedical data. The asymmetry between the tens of thousands of publications describing novel biomarkers, and the tiny fraction of those that ultimately become clinical diagnostic tests, as noted in 2014, has persisted, representing a profound failure of biomarker discovery.2,3 Why?
The torrent of data enabled by technical advances and digitization led to a 2008 conclusion that the traditional scientific method needed to be refined, as standard hypothesis testing was not compatible with the availability of “big data”.41 Computers analyzing large datasets can yield important previously undetectable correlations as seen in astronomy, cosmology and meteorology.42,43 However, in biomedical research, this approach has yielded an underwhelming dividend, as illustrated by the poverty of novel clinically useful biomarkers.
We hypothesized that prior analytic approaches have not been able to reveal useful biomarkers, because of their lack of suitability for analyzing complex biomedical data, neglecting biology and disease as complex systems.3
Our software solution differs from the various types of machine learning, by fusing evolutionary computation with algorithmic information theory, incorporating a wide range of mathematical functions to directly address the non-linearity and high dimensionality of complex biomedical data yielding transparent quantitative predictive algorithms. Transparency is necessary for both informing medical decisions and providing scientific insight,14,15,44 for patients, physicians and regulators, and also to provide a basis for the development of clinical tests.
The software automatically eliminates overfitted algorithms from consideration. Confirmation that the selected algorithm is not overfitted is done in the third stage of the automated process. The application of concepts from evolutionary biology - inheritance, random variation, and natural selection - explains why evolutionary computation can handle large high dimensional data so efficiently.45 Information theory is a mathematical framework for understanding information at a fundamental level, which includes the concepts of randomness and algorithmic compression.46,47 That nature, in all its manifestations, is algorithmic, and that scientific comprehension is a process of finding predictive algorithms that compress information into its essence, is the basis for our approach.47–49 Our algorithmic focus is complemented by the emerging understanding that biology reflects molecular networks that are algorithmic.50 Biological functions, including therapeutic response and disease are recognized to be mediated by molecular networks, variously defined as modules, motifs or cores.5,51–54 It has been proposed that understanding of molecular networks will be necessary for a deeper understanding of biological information flow and that this will require an algorithmic framework.5,50 The incorporation of many mathematical functions into the software allows the underlying molecular interactions, which are frequently non-linear, to be modeled in the selected algorithms, thereby maximizing their predictivity, without any a priori assumptions as to their nature. The incorporation of mathematical functions into the algorithms aligns with emerging perspectives around the need to incorporate mathematics into biology.55
We posit that our approach is practical and effective with modest sample sizes because of its focus on defining the most salient signals, as represented by the mathematical relationships between variables, which is predominantlywhere the information resides in complex systems. The evolutionary foundation of our approach allows large numbers of variables to be analyzed without the need to use arbitrary thresholds and feature selection to eliminate variables from consideration. The number of potential permutations of algorithmic memory registers containing only the final eight variables, combined with available mathematical functions in the analysis we report, is in excess of 1061. When considering all possible 52,379 biomarkers in the discovery dataset for each patient, the treatment arm variable, the software’s complete palette of available mathematical functions, and a limit of only 16 instructions per algorithm, the total number of potential discovery algorithms is 4.749 times 101253.
The discovery algorithm that we present incorporates the sine function, showing that molecular interactions underlying the response to therapy in RA can be represented using mathematical functions that also depict relationships in other biological contexts. This implies that the algorithm depicts a fundamental phenomenon.
We identify lower expression variables that are likely to have disproportionate biological effects because of their non-linear interactions that other analytic approaches cannot readily detect and have frequently been excluded from consideration. These variables, which are not well studied, are likely to provide insights into the molecular mechanisms underlying responsiveness to anti-TNF therapy in RA. Individually, the eight variables were not highly correlated with response, and would not be useful response predictors, either individually or collectively, in the absence of the mathematical components of the algorithm.
As well as revealing predictive algorithmic biomarkers, our approach may also enable the identification and understanding of the molecular networks mediating disease and therapeutic effects. These algorithms may help with the identification and prioritization of novel therapeutic targets, particularly those that might be amenable to emerging approaches that modulate specific RNA transcripts. The accuracy we report defines three distinct patient subsets: the first are anti-TNF responders, the second are placebo responders and the third are non-responders to either anti-TNF therapy or to placebo. The potential to predict patients in these three categories will be a focus of future studies to inform the most appropriate individualized therapeutic interventions.
There are no prior reports directly linking SPTY2D1, KCTD4, and c1orf105 to either RA, immune-mediated diseases or to responses to therapy.24–28 TPM3 has been reported as an autoantigen in RA, and to be associated with both myopathy and renal cancer.33–35 ARHGDIB expression is upregulated in RA, whereas SIAH1 is downregulated.36–38 TPM3, ARHGDIB and SIAH1 have not been associated with therapeutic outcomes. Prior cytomegalovirus exposure has been associated with poor responses to therapy in early RA29,30 which may explain the UL84 gene variable association as it encodes for a cytomegalovirus protein. Mutations in PTPRC, also known as CD45, have been associated with RA patient response to anti-TNF therapy.31,32
The discovery variables we report and have validated, are in development as components of a clinical PCR test using clinical grade instruments and protocols. PCR tests are versatile and reliable for deployment in clinical laboratories. The discovery algorithm, derived on research instruments such as we have presented here, is not appropriate for clinical use, as its sole purpose is to identify the most informative variables. Research-stage instruments have different quantitation levels, dynamic range and reliability compared to clinical-grade assay processes and technology. Therefore, the clinical test we have in development will require derivation of novel algorithms, which will then need to be prospectively validated for clinical use.
We envision that the clinical test will provide the basis for an ensemble of algorithms, each predictive of different specific clinical endpoints, in addition to DAS28, at different time points, thereby providing a comprehensive efficacy profile for individual patients, most informative to clinicians, to maximize clinical relevance.
We are applying the same gene expression variables to other diseases responsive to anti-TNF therapy. Our hypothesis is that the same variables will be the basis of algorithms predictive of individual treatment response to anti-TNF therapies in other diseases where anti-TNF therapy is of proven efficacy, implying that the variables we have identified are of fundamental biological importance in mediating the efficacy of anti-TNF therapy in general.
Our analytic approach may provide a novel paradigm for future synchronous development of novel therapies and companion diagnostics. Proof of concept clinical studies could routinely incorporate baseline biomarker profiling to yield transparent algorithms predictive of both efficacy and safety signals. The predictive algorithmic biomarkers could then be expeditiously incorporated into and validated in subsequent registration studies, potentially yielding highly predictive companion diagnostics to inform both regulatory approval and reimbursement decisions. Such algorithmic biomarkers could also be incorporated into the product label to inform prescribing decisions for individual patients.
In the future, we envision all therapies could be accompanied by algorithmic biomarkers, predictive of efficacy and safety endpoints of interest, to objectively and quantitatively inform administration decisions for individual patients. The algorithmic biomarkers could be promptly updated as new data emerge to optimize clinical utility. In addition, clinical trial outcomes could be routinely summarized in terms of the algorithms.
Optimal translation of biomedical data into actionable information requires analytic methodology designed to yield mathematically informed algorithmic insight from complex high dimensional non-linear data. The novel analytic approach that we present addresses that challenge. This is the only reported method that provides transparent, simple algorithmic biomarkers that accurately reflect biology as a complex system and quantitatively predicts individual therapeutic responses, that can readily be translated into clinical tests. This may have implications for the discovery and development of companion diagnostics and the analysis of clinical trials.
Currently, the gulf between actual health care and that which could be provided, has arguably never been wider.56 We contend that bridging that gulf will require solving the “biomarker problem” with biomarkers that reliably identify those patients who are most likely to benefit from a particular agent.57 As we have shown here, this will be done by a focus on medicine as an information science, acknowledging that “to be useful, data must be analyzed, interpreted, and acted on. Thus, it is algorithms, not data sets, that will prove transformative.”58
The study that provided the discovery data was conducted in accordance with principles of Good Clinical Practice and was approved by the National Ethics Committee in Romania and the National Ethics Committee, Clinical Research of Drugs and Methods of Treatment in Moldova, with all subjects providing informed written consent.16,17 Written informed consent for all participants and local ethics approval was obtained for each of the studies that provided validation data as detailed in the original publications.18–23
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)