Keywords
Keywords: Neuroimmune Crosstalk, Chronic Stress, HPA Axis, Neuroinflammation, Precision Medicine
Keywords: Neuroimmune Crosstalk, Chronic Stress, HPA Axis, Neuroinflammation, Precision Medicine
Stress is one of the most widespread health determinants globally, affecting an estimated 70-90% of adults at some point in their lives. Short-term stress can enhance survival by mobilizing energy and immune defenses, prolonged or chronic stress has been associated with poor health outcomes (Piao et al., 2024). According to the World Health Organization, stress-related mental disorders such as depression and anxiety affect more than 970 million people worldwide (Al-Garni et al., 2025). The economic burden of stress-related disorders is estimated to exceed US$1 trillion annually in lost productivity. Beyond mental health, chronic stress contributes to systemic diseases, including cardiovascular disease, diabetes, autoimmune disorders, and cancer, making it a major global public health concern. Stress responses are coordinated through neuroimmune interactions that link the brain and the immune system (Kavvadas et al., 2023). These interactions involve the hypothalamic-pituitary-adrenal (HPA) axis, the sympathetic-adrenal-medullary (SAM) system, and peripheral immune circuits. Collectively, they regulate the release of stress hormones such as cortisol and adrenaline, along with immune messengers like cytokines, to maintain homeostasis. However, sustained activation of these pathways has been reported to disrupt their regulatory functions (Mbiydzenyuy & Qulu, 2024). Elevated inflammatory cytokines such as IL-6, TNF-α, and IL-1β are observed in patients with major depressive disorder (MDD) and post-traumatic stress disorder (PTSD). This has been reported using positron emission tomography (PET) imaging, which consistently reveals glial activation in these populations (Min et al., 2023). In Alzheimer’s disease, chronic microglial activation contributes to impaired clearance of amyloid-β and accelerates neuronal degeneration (Min et al., 2023). Similar patterns are reported in Parkinson’s disease and other neurodegenerative conditions. Chronic stress-related inflammation increases the risk of coronary artery disease by 40% and is strongly linked to insulin resistance and metabolic syndrome (Valiukas et al., 2025). Despite progress, important research gaps remain why some individuals exposed to prolonged stress develop severe psychiatric or neurodegenerative disorders while others maintain resilience (James et al., 2023). The contribution of sex-specific biology, early-life adversity, and gut microbiome diversity to these outcomes is still poorly characterized. Moreover, although mechanistic studies have reported the roles of glucocorticoid receptor resistance, persistent cytokine signaling, and epigenetic modifications, translation into effective therapies has been limited (Beurel & Nemeroff, 2024). Biomarker-driven patient stratification, longitudinal studies, and integrative multi-omics approaches are urgently needed to bridge this gap. This review synthesizes current evidence on neuroimmune interactions in stress, focusing on physiological systems, molecular mechanisms, and health implications.
The HPA axis and SAM are responsible for coordinating both hormonal and neural outputs that prepare the body to respond to stress and regulate long-term immune function. These responses are adaptive under acute stress. However, chronic activation disrupts their regulatory balance, leading to persistent immune dysfunction and increased vulnerability to disease as discussed in sub-sequent sections.
The HPA axis coordinate neuroendocrine signaling that leads to cortisol release and systemic adaptation to physiological and psychological challenges. Stress signals from amygdala and prefrontal cortex stimulate the hypothalamus to release corticotropin-releasing hormone (CRH) and vasopressin (Mbiydzenyuy & Qulu, 2024). These hormones activate the pituitary gland, which secretes adrenocorticotropic hormone (ACTH). ACTH stimulates cortisol release from the adrenal cortex, which inhibit inflammation by binding to glucocorticoid receptors in immune cells and suppressing pro-inflammatory gene expression (Allen & Sharma, 2023). In acute stress, this mechanism prevents excessive inflammation and promotes recovery. In chronic stress, elevations of cortisol lead to glucocorticoid resistance, a condition where immune cells no longer respond effectively to cortisol’s anti-inflammatory signals (Sic et al., 2024). Evidence from patients with major depressive disorder have shown elevated cortisol alongside increased inflammatory cytokines such as IL-6 and TNF-α (Hassamal, 2023). Epigenetic studies further reveal that individuals exposed to early-life adversity often show changes in the NR3C1 gene (which encodes the glucocorticoid receptor), making them more vulnerable to stress-induced inflammation in adulthood. This alteration reduces receptor expression and contributes to impaired stress regulation throughout life, increasing susceptibility to psychiatric disorders and chronic inflammatory diseases (Forum et al., 2025).
The SAM system complements the HPA axis by mediating the rapid, “fight-or-flight” response. Activation of sympathetic nerves triggers the release of adrenaline and noradrenaline, which act on adrenergic receptors in multiple tissues, including immune cells. This signaling affects immune processes such as chemotaxis, phagocytosis, and cytokine release (Feng et al., 2025). Activation of β2-adrenergic receptors has been shown to suppress inflammation through increased IL-10 production, whereas α-adrenergic signaling has more recently been associated with the amplification of pro-inflammatory activity (Damo et al., 2023). Sustained sympathetic tone leads to reduced natural killer (NK) cell cytotoxicity, impaired lymphocyte proliferation, and eventual immune exhaustion. For example, caregivers of patients with Alzheimer’s disease, who experience long-term psychosocial stress, exhibit reduced NK cell activity and weaker antibody responses to vaccines compared with non-caregivers (Madison et al., 2021). Similarly, occupational stress has been linked to higher rates of respiratory infections and slower wound healing, consistent with the suppressive effects of prolonged adrenergic signaling on adaptive immunity (Alotiby, 2024). Additionally, Persistent SAM overactivation also contributes to neuroimmune dysfunction. Elevated noradrenaline in the central nervous system can increase blood-brain barrier permeability and stimulate microglial activation and neuroinflammation (Obeagu, 2025).
The timing and duration of stress exposure are important in determining whether stress acts as an immune enhancer or suppressor. Acute stress, typically lasting minutes to a few hours, generally enhances immune readiness. Short-term activation of the HPA axis and SAM system mobilizes innate immune cells such as neutrophils and monocytes into circulation (Albitar et al., 2025). This stimulates the release of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-6 (IL-6). These changes prepare the body to mount defense against potential injury or infection. Clinically, controlled laboratory studies have shown that acute stress before vaccination can enhance antibody production, suggesting a beneficial role for brief stress exposures in priming immune responses (Al-Qahtani et al., 2024). Chronic stress, in contrast, stimulates prolonged activation of the HPA axis, leading to disruption of normal cortisol rhythms and reduced sensitivity of glucocorticoid receptors. Simultaneously, sustained sympathetic activation maintains elevated catecholamine levels, which exhaust immune cell function over time. The combined effect is suppression of adaptive immunity characterized by reduced T- and B-cell proliferation alongside persistent, low-grade inflammation (Mikolaskova et al., 2024). A prospective study in medical students showed that exam stress slowed wound healing, demonstrating how even moderate stress can impair recovery (Mochel et al., 2025). Likewise, trauma survivors often display elevated inflammation linked to poorer health outcomes. Table 1 provides a comparative overview of the immunological, neuroendocrine, and systemic effects of acute versus chronic stress. This indicates the transition from short-term adaptive responses to long-term pathological outcomes.
Stress signaling through the HPA and SAM systems has direct effect om the central nervous system. Glucocorticoids and catecholamines influence the permeability of the BBB, allowing peripheral immune cells and cytokines to access the brain parenchyma. This process facilitates the activation of microglia, the CNS’s resident immune cells, which, upon chronic stimulation, adopt a pro-inflammatory phenotype (Albitar et al., 2025). These activated microglia release neurotoxic mediators such as IL-1β, TNF-α, and reactive oxygen species (ROS), contributing to neuroinflammation, synaptic dysfunction, and neuronal injury. Such neuroimmune activation has been observed in rodent models of chronic mild stress and has been linked to the development of depression-like behaviors and cognitive deficits (Woodburn et al., 2021). Furthermore, peripheral immune molecules influenced by chronic stress can serve as neuromodulators within the brain. Cytokines like IL-1β and IL-6 affect synaptic transmission, neurogenesis, and neurotransmitter metabolism, particularly in stress-sensitive brain regions such as the hippocampus and prefrontal cortex (Knezevic et al., 2023). For example, elevated IL-6 levels in cerebrospinal fluid have been correlated with memory impairment and increased risk for Alzheimer’s disease (Woodburn et al., 2021). The complex interplay between psychological stress and immune function is mediated through two primary neuroendocrine circuits, the HPA axis and the SAM system, as depicted in Figure 1.
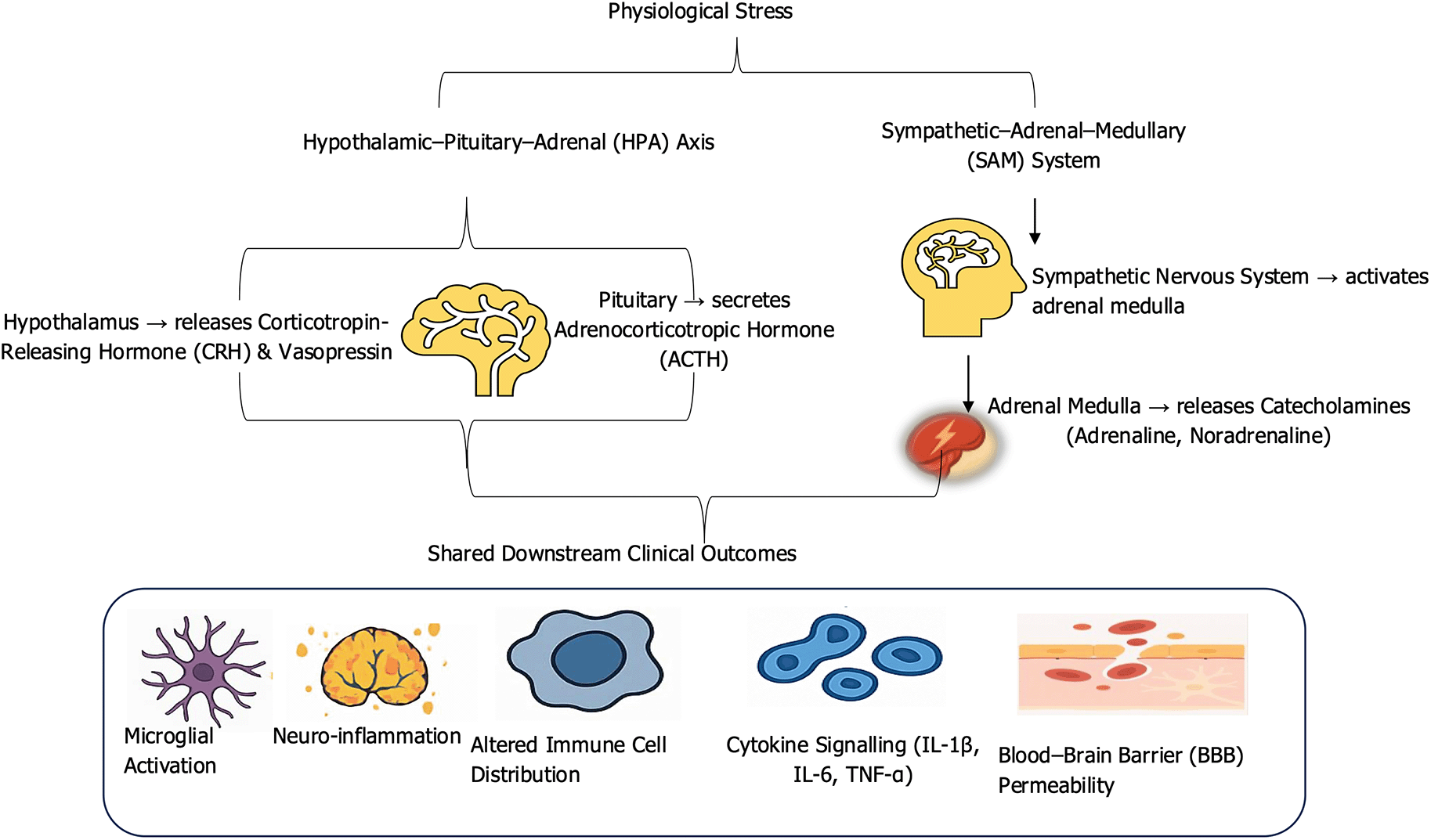
The schematic depicts how stress response systems stimulate inflammatory processes via glucocorticoid release, whereas the sympathetic–adrenal–medullary (SAM) system rapidly mobilizes catecholamines that act on adrenergic receptors of immune cells. Convergence of these pathways alters blood–brain barrier integrity, promotes neuroimmune crosstalk, and drives neuroinflammation, increasing vulnerability to stress-related disorders.
The nervous and immune systems communicate through complex molecular pathways that regulate both homeostasis and stress responses, as depicted in Figure 2. These mediators enable adaptive coordination during acute stress and drive maladaptive neural and immune changes under chronic stress. Key mechanisms include glucocorticoid receptor signaling, cytokine pathways, microglial activation, epigenetic reprogramming, and neuropeptide-mediated modulation of immune tone, as outlined in the following subsections.
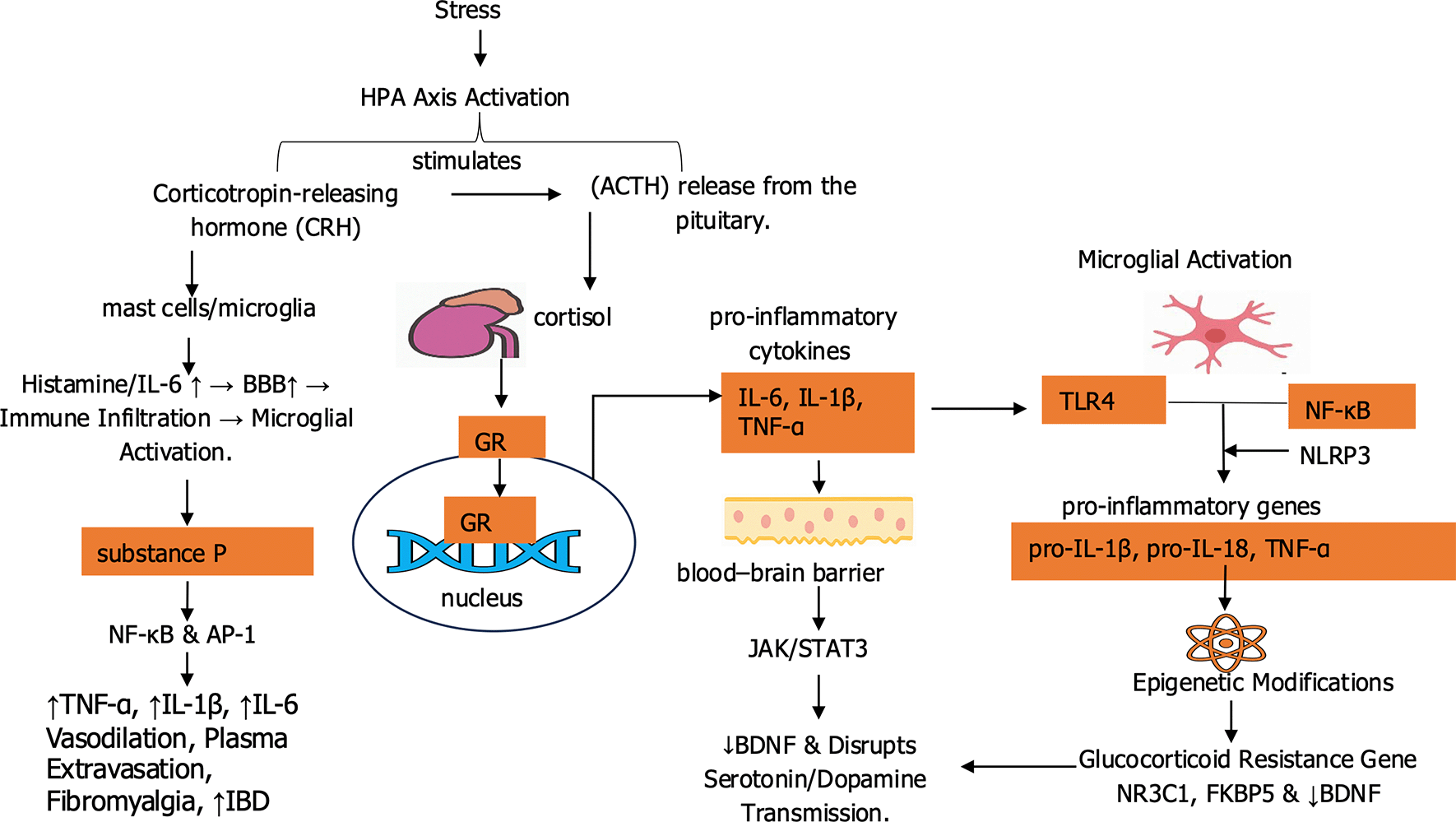
This figure illustrates how stress mediators activate both endocrine and immune pathways. Substance P and glucocorticoids act through glucocorticoid receptors to influence gene transcription, whereas immune activation via TLR4 and NF-κB signaling drives the release of pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, pro-IL-1β, and pro-IL-18. Together, these processes link stress to chronic neuroinflammation and altered neural activity.
Glucocorticoid receptor (GR) signaling is a central pathway linking stress responses with immune and neural function. Cortisol is released following the activation of the HPA axis, which binds to GRs expressed in immune and neural cells. The ligand–receptor complex translocates to the nucleus, where it interacts with glucocorticoid response elements (GREs) to regulate gene transcription (Knezevic et al., 2023). In immune cells, GRs suppress inflammation through trans-repression, inhibiting transcription factors such as NF-κB and AP-1 and reducing cytokine production (IL-6, IL-1β, TNF-α). This mechanism explains the clinical effectiveness of synthetic glucocorticoids like dexamethasone, though the same immunosuppressive action can increase infection risk, as seen in COVID-19 patients receiving steroid therapy (Zhao et al., 2024). In the brain, GRs modulate synaptic plasticity, stress regulation, and cognition, particularly in the hippocampus and prefrontal cortex. Evidence from both animal and human studies links glucocorticoid resistance to major depressive disorder (MDD) and post-traumatic stress disorder (PTSD), where systemic inflammation coexists with HPA axis dysregulation (Misiak et al., 2020). Some studies suggest immune cells are more affected than neural tissue, while others implicate epigenetic changes. For example, hypermethylation of the GR gene (NR3C1) following early-life trauma is associated with reduced GR expression and increased vulnerability to stress-related disorders (Hatta et al., 2025).
Cytokines represent an important interface between the immune system and the brain, functioning as both immune messengers and neuromodulators. Stress stimulates the secretion of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α in both the periphery and the central nervous system (CNS) (Lesovaya et al., 2022). These molecules access neural targets by crossing vulnerable regions of the BBB, using transport mechanisms, or signaling indirectly via vagal afferents. They activate intracellular cascades such as the JAK/STAT pathway, particularly STAT3, which alters neuronal excitability and reduces synaptic plasticity (T. Chen et al., 2024). In rodents, IL-6 administration impairs long-term potentiation (LTP), the cellular basis of learning and memory, while IL-1β infusion suppresses hippocampal neurogenesis (Leschik et al., 2021). Similar claims have been reported in human studies linking systemic inflammation with cognitive dysfunction in mood and anxiety disorders (Mac Giollabhui et al., 2024). Elevated cytokines further reduce brain-derived neurotrophic factor (BDNF) expression and disrupt monoamine neurotransmission, particularly serotonin and dopamine, creating a neurochemical profile resembling depression (Cui et al., 2024). Stress-induced cytokine signaling contributes to neurochemical dysregulation by impairing synaptic plasticity, reducing neurotrophic support, and disrupting neurotransmitter balance. This provides a mechanistic link between peripheral immune activation and central emotional regulation, reinforcing the role of inflammation as amplifier of mood and stress-related disorders (Osimo et al., 2020).
The activation of microglia remains the central feature of stress-related neuroimmune dysfunction is the brain’s resident immune cells. Microglia act as frontline defenders in the central nervous system and respond quickly to stress signals. An early step in this response is the activation of Toll-like receptor 4 (TLR4) by stress-related danger signals. This triggers NF-κB signaling and increases the production of inflammatory mediators such as inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and cytokines (Elbakary et al., 2025). This inflammatory response can be amplified by the NLRP3 inflammasome, a protein complex that activates caspase-1 and converts pro-IL-1β and pro-IL-18 into their active forms. The NLRP3 inflammasome is especially sensitive to stress-related cellular changes, including increased reactive oxygen species (ROS), ATP release, and mitochondrial dysfunction. Evidence from animal studies reported that rodents exposed to chronic stress show greater microglial activation, higher hippocampal IL-1β levels, and depressive-like behaviors, which can be reduced by blocking or deleting NLRP3 components (Shi et al., 2023). Chronically active microglia promote excessive synaptic pruning, dendritic spine loss, and reduced neurogenesis, leading to cognitive decline and emotional dysregulation similar to Alzheimer’s disease and psychiatric disorders (Wang et al., 2023).
Stress exposure, especially during sensitive developmental periods such as early childhood or adolescence, induces long-term modifications in gene expression through epigenetic mechanisms (Shi et al., 2023). These include DNA methylation, histone modifications, and the regulation of gene expression by non-coding RNAs. Hypermethylation of the NR3C1 gene promoter region has been observed in individuals exposed to childhood trauma (Cicchetti & Handley, 2017). This results to reduced GR expression and impaired feedback inhibition of the HPA axis. The alteration enhances glucocorticoid resistance, prolonging elevated cortisol and increased inflammatory signaling. Another regulator is FKBP5, a co-chaperone of the GR complex that modulates receptor sensitivity (Hatta et al., 2025). FKBP5 expression is increased by stress and shows stress-related hypomethylation in patients with PTSD and major depression (Buttgereit et al., 2025). Animal studies provide evidence with maternal separation in rodents increases DNA methylation at the BDNF gene promoter, reducing neurotrophic factor expression and impairing neuronal resilience (X. Zhang et al., 2020). This epigenetic downregulation of BDNF compromises neuroplasticity and increases vulnerability to depressive-like behaviors in adulthood. These epigenetic markers are recognized as mediators of long-term risk for psychiatric disorders such as depression, anxiety, and PTSD, as well as somatic conditions including cardiovascular disease and chronic pain syndromes. Interventions such as psychotherapy, antidepressants, physical activity, and even targeted epigenetic drugs (e.g., histone deacetylase inhibitors) have shown promise in modulating stress-related epigenetic patterns, suggesting that maladaptive programming may be at least partly corrected (Lu et al., 2025).
Neuropeptides also play a fundamental role in bridging the nervous and immune systems. Among the most prominent are corticotropin-releasing hormone (CRH), substance P, and neuropeptide Y (NPY), which have direct effects on immune cell activity and inflammation (Yeo et al., 2022). CRH, besides its central function in initiating the HPA axis cascade, can bind to receptors on mast cells, microglia, and peripheral immune cells, promoting the release of IL-6, histamine, and other pro-inflammatory mediators. Substance P, acting via neurokinin-1 (NK1) receptors, stimulates macrophage activation and enhances cytokine release, contributing to conditions such as neurogenic inflammation, fibromyalgia, and inflammatory bowel disease all of which are increased by chronic stress (Lauritano et al., 2023). In contrast, neuropeptide Y (NPY) generally exerts anti-inflammatory effects, promoting the production of IL-10 while reducing TNF-α levels. NPY also inhibits microglial activation and has been associated with greater resilience to stress in both animal models and human cohorts (W. C. Chen et al., 2020).
Cytokines form an important link between psychological stress and systemic inflammation through tightly regulated feedback loops. Acute stress activates HPA axis and SAM system, releasing cortisol and catecholamines. These hormones normally suppress excessive inflammation and restore balance. Under chronic or repeated stress, immune cells develop reduced sensitivity to glucocorticoids resulting to glucocorticoid resistance. This breakdown in negative feedback allows persistent production of pro-inflammatory cytokines, particularly interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) (Auger et al., 2024). These cytokines can penetrate the blood–brain barrier via active transport or signal indirectly through endothelial and vagal pathways. Once within the central nervous system, they further stimulate the hypothalamus and brainstem, driving additional HPA and SAM activity (Krasner et al., 2025). The result is a self-repeating cycle in which stress hormones and cytokines reinforce one another. Adaptive in the short-term enhancing immune vigilance and energy mobilization this feedback becomes maladaptive when prolonged. Sustained cytokine activity maintains systemic inflammation, contributing to cardiovascular disease, metabolic syndrome, and neuropsychiatric disorders. These feedback loops as depicted in Figure 3 illustrate how stress responses, initially protective, can transform into amplifier of chronic disease (Valiukas et al., 2025).
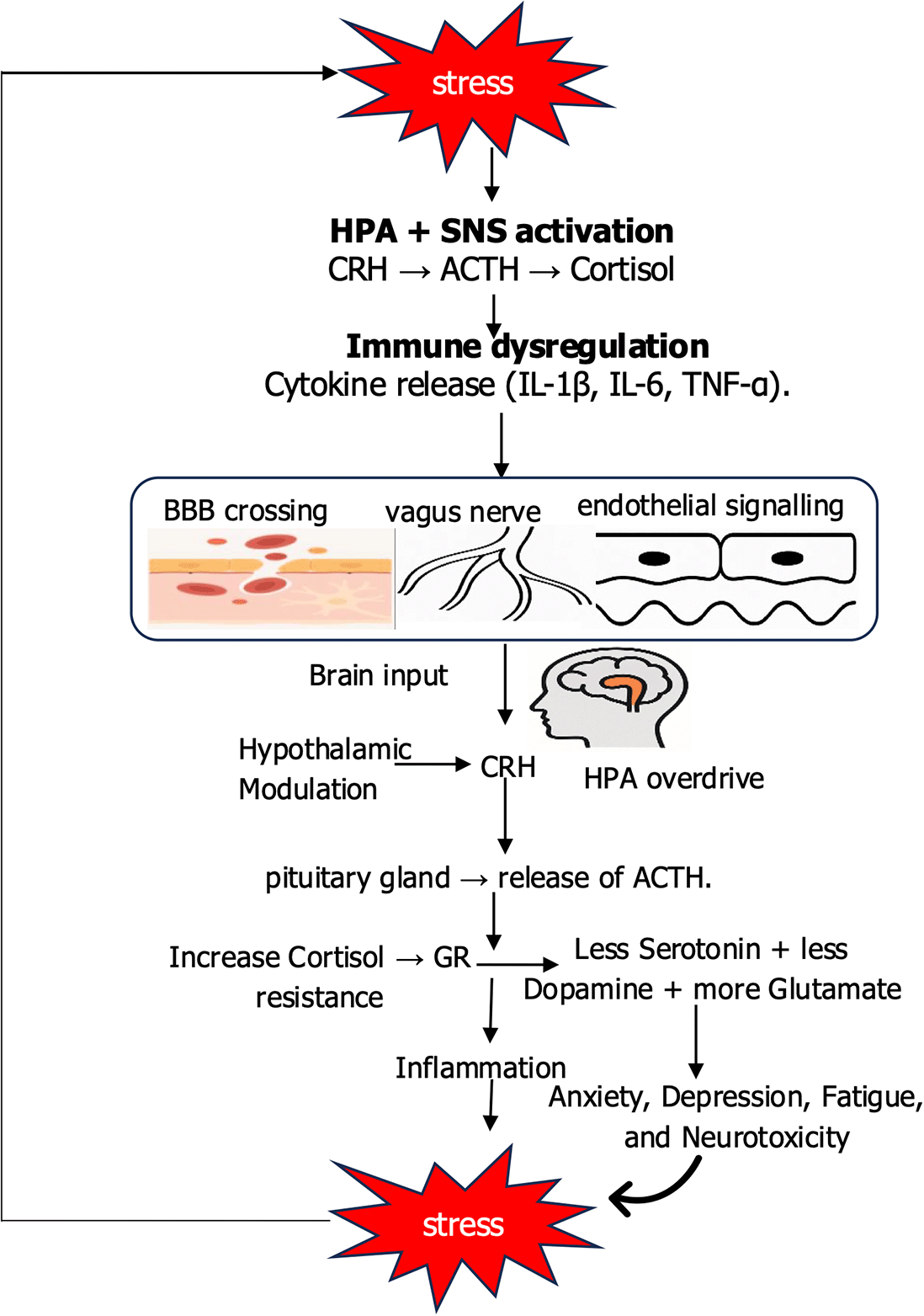
The figure illustrates how stress initiates peripheral immune activation, leading to cytokine release, vascular and neuronal changes, and blood–brain barrier disruption. These signals feed back into central brain circuits, particularly the hypothalamus, reinforcing stress responses and establishing a self-amplifying cycle that drives neurophysiological dysfunction.
The persistent activation of the HPA axis and the sympathetic nervous system (SNS) under chronic stress leads to disruption in immune homeostasis. This neuroimmune dysregulation is characterized by a dual-edged immune profile, diminished antiviral and cytotoxic immune surveillance on one hand, and sustained pro-inflammatory signaling on the other. This imbalance increases vulnerability to infections, autoimmunity, neuropsychiatric disorders, cancer progression, and cardiometabolic diseases, as discussed subsequently.
Chronic stress compromises both innate and adaptive immunity, particularly impairing early antiviral defenses. The type I and II interferon (IFN) pathways, essential for detecting and containing viral infections, are suppressed under prolonged SNS and HPA activation (Lauritano et al., 2023). Catecholamines and cortisol reduce IFN-α/β production in plasmacytoid dendritic cells (pDCs), impairing the initial antiviral response. Concurrently, natural killer (NK) cell cytotoxicity, which is important in clearing virus-infected and transformed cells, depleted. Glucocorticoids downregulate MHC-II and co-stimulatory molecules (e.g., CD80/CD86), reducing T-cell priming efficiency (Troise et al., 2024). Clinically, stressed individuals exhibit weaker and shorter-lived antibody responses following vaccination. A longitudinal study of medical students during exam periods found significantly lower antibody titers post-influenza vaccination, with the effect most pronounced in those reporting high perceived stress. Similarly, older adults under chronic stress show reduced seroconversion rates to the pneumococcal and shingles vaccines (Ehrlich et al., 2024). Additionally, stress disrupts epithelial integrity, with cortisol and norepinephrine reducing the expression of tight junction proteins (e.g., occludin, ZO-1) and suppressing antimicrobial peptides like defensins. This increases susceptibility to respiratory and gastrointestinal pathogens and explains why individuals under high psychosocial strain report more frequent upper respiratory infections (Huang et al., 2025). In chronic viral infections like HIV, stress enhances viral replication and impairs immune response. Catecholamines activate the cAMP/PKA pathway, which stimulates HIV long terminal repeat (LTR), resulting in increased viral transcription. Simultaneously, stress-induced suppression of CD8+ T-cell function and NK activity reduces viral clearance. Longitudinal cohort studies (e.g., the Multicenter AIDS Cohort Study) show that HIV-positive individuals with high perceived stress or depression progress to AIDS more rapidly, independent of antiretroviral therapy adherence (Zeleke et al., 2025).
In autoimmune diseases, stress rarely acts as a primary trigger but functions as a potent disease amplifier that increases the burden. Persistent SNS activation has been reported to promote pro-inflammatory immune phenotype expression. β2-adrenergic signaling in immune cells enhances production of IL-6, IL-1β, and TNF-α in myeloid cells and suppressed anti-inflammatory IL-10 (Sah & Singewald, 2025). This increased glucocorticoid resistance, commonly observed in patients with rheumatoid arthritis (RA) and lupus, where immune cells fail to respond to cortisol’s anti-inflammatory signals due to reduced glucocorticoid receptor (GR) expression or altered GR translocation (Lockett et al., 2024). Furthermore, chronic stress promotes Th17 differentiation, a pro-inflammatory T-helper subset implicated in tissue damage by IL-6 and TGF-β signaling, which simultaneously impairs regulatory T-cell (Treg) function, reducing immune tolerance. In multiple sclerosis (MS), stress is linked to increased relapse rates. A prospective study found that MS patients experiencing major life events had a higher risk of relapse in the following 6–8 weeks, likely due to stress-induced blood–brain barrier (BBB) disruption and enhanced leukocyte trafficking (Dziedzic et al., 2024). In rheumatoid arthritis (RA), stress correlates with increased disease activity, pain, and fatigue. Functional MRI studies show that RA patients under stress exhibit increased amygdala activation and reduced prefrontal regulation linking emotional processing to peripheral inflammation. Moreover, stress-induced cortisol flattening (loss of diurnal rhythm) is associated with higher CRP and joint swelling (Dziedzic et al., 2024).
Chronic stress induces a neuroinflammatory state that contributes directly to the pathophysiology of depression, anxiety, and PTSD. Peripheral cytokines such as IL-6, IL-1β, and TNF-α signal the brain via multiple routes, including active transport across the blood–brain barrier (BBB), binding to endothelial receptors, and vagal afferent activation. These signals activate microglia, the brain’s resident immune cells (Huang et al., 2025). This leads to excessive synaptic pruning, reduced neurogenesis (particularly in the hippocampus), and altered connectivity in cortico-limbic circuits governing mood, fear, and executive function. A key metabolic pathway linking inflammation to depression is the kynurenine pathway of tryptophan metabolism. Inflammatory cytokines (especially IFN-γ and IL-6) upregulate the enzyme IDO (indoleamine 2,3-dioxygenase), shifting tryptophan away from serotonin synthesis and toward kynurenine (Roberts et al., 2022). Kynurenine metabolites like quinolinic acid are NMDA receptor agonists that promote excitotoxicity and oxidative stress, while kynurenic acid may impair glutamate clearance. This dual action disrupts monoaminergic and glutamatergic signaling core mechanisms in depression. Clinically, patients with treatment-resistant depression have been reported to exhibit elevated CRP and IL-6. Moreover, interferon-α therapy for hepatitis C induces depressive symptoms in 30–50% of patients, reversible with antidepressants or anti-inflammatory agents (J. P. C. Chang et al., 2025).
Although stress does not initiate cancer, it chronically activates the sympathetic–adrenomedullary system and the HPA axis, increasing the release of catecholamines from sympathetic nerve terminals and the adrenal medulla and elevating cortisol. These mediators accumulate within the tumor microenvironment and bind β-adrenergic and glucocorticoid receptors on tumor, endothelial, fibroblast, and immune cells, triggering cAMP–PKA/CREB, MAPK, and STAT3/NF-κB pathways (Mujinya et al., 2025). The result is upregulation of VEGF and matrix metalloproteinases, induction of epithelial–mesenchymal transition, enhanced angiogenesis, and suppression of antitumor immunity (reduced NK/CD8+ T-cell activity with expansion of myeloid-derived suppressor cells and Tregs), accelerating invasion and metastasis. In ovarian and breast cancer models, social isolation stress increases metastatic burden by 30–50%, an effect blocked by β-blockers like propranolol (Khan et al., 2025). Additionally, stress reduces dendritic cell maturation, NK cell cytotoxicity, and CD8+ T-cell infiltration. It also increases myeloid-derived suppressor cells (MDSCs) and Tregs, creating an immunosuppressive tumor microenvironment. This shift is toxic during perioperative periods, when surgical stress and anesthesia amplify catecholamine release, potentially promoting micrometastatic seeding (Roberts et al., 2022). Retrospective studies suggest that patients taking β-blockers during breast cancer surgery have lower recurrence rates and improved survival. A meta-analysis supplemented this by reporting a significant association between chronic stress and increased cancer mortality, particularly for breast and prostate cancers (Caparica et al., 2021). However, confounding factors (e.g., lifestyle, access to care) complicate interpretation. Prospective trials testing perioperative β-blockade (e.g., the ongoing POISE-3 ancillary study) can clarify causality. Future research should explore biomarker-guided interventions, including β-blockers, mindfulness-based therapies, and autonomic modulation, in high-risk cancer populations (McKenzie et al., 2023).
Chronic stress is a recognized risk factor for metabolic syndrome, type 2 diabetes, and cardiovascular disease, acting through interconnected neuroendocrine, inflammatory, and behavioral pathways. Sustained SNS activation increases heart rate, vasoconstriction, and renin release, contributing to hypertension and endothelial dysfunction. Norepinephrine reduces nitric oxide (NO) bioavailability, promoting vasoconstriction and platelet aggregation (Dziedzic et al., 2024). Similarly, prolonged cortisol exposure increases hepatic gluconeogenesis, promotes central adiposity, and reduces insulin sensitivity in skeletal muscle and adipose tissue, major features of the metabolic syndrome (Bavaresco et al., 2024). These neuroendocrine effects are reinforced by low-grade inflammatory signaling and stress-linked behaviors (e.g., sleep disruption, physical inactivity, and adverse dietary patterns), creating a self-reinforcing milieu that accelerates the development of cardiometabolic disease (Khan et al., 2025). Stress-induced cytokines (IL-6, TNF-α) upregulate endothelial adhesion molecules (VCAM-1, ICAM-1), promoting monocyte recruitment into the arterial wall. These differentiate into lipid-laden foam cells, forming atherosclerotic plaques (Krasner et al., 2025). Oxidative stress from catecholamine auto-oxidation further damages vascular tissue. The Whitehall II study found that employees with high job strain had a 50% higher risk of coronary heart disease over 10 years, independent of traditional risk factors (Z. Zhang et al., 2023). Similarly, the INTERHEART study identified psychosocial stress as one of the top modifiable risk factors for myocardial infarction globally. Stress acts as a risk multiplier that lowers the threshold for disease in genetically or metabolically vulnerable individuals. Interventions that target both stress (e.g., mindfulness) and lifestyle (e.g., exercise, diet) are likely to have synergistic benefits, as demonstrated in cardiac rehabilitation programs (Javila et al., 2025).
Advances in understanding stress-induced neuroimmune dysregulation have shifted therapeutic strategies away from symptom suppression toward targeted modulation of the underlying pathways. This is achieved by identifying molecular and cellular mechanisms that sustain chronic neuroinflammation as discussed in subsequent sections. Several promising therapeutic targets have emerged across glucocorticoid signaling, cytokine modulation, microglial activity, inflammasome regulation, and epigenetic reprogramming.
Pharmacological agents that modulate stress-related signaling pathways have demonstrated potential in reducing systemic inflammation, preventing tumor progression, and restoring hormonal and immune balance. These agents include β-blockers, glucocorticoid receptor modulators, and cytokine inhibitors. β-adrenergic receptor antagonists, such as propranolol, inhibit stress-induced tumor progression by blocking the effects of norepinephrine on β2-adrenergic receptors expressed on tumor and immune cells (McKenzie et al., 2023). In preclinical models of breast cancer, β-blockers reduce metastasis by suppressing tumor-associated inflammation and angiogenesis. This has been reported to improve survival in cancer patients using β-blockers; however, randomized controlled trials are still needed to confirm causality (Sharma et al., 2025). Additionally, the heterogeneity of β-receptor expression across tumor types and the potential for off-target effects remain important considerations. Glucocorticoid receptor (GR) modulators aim to address glucocorticoid resistance, a common consequence of chronic stress in which immune cells become less responsive to cortisol, leading to unchecked inflammation (A. Chang et al., 2023). Compounds such as mifepristone (RU-486) and newer selective GR modulators like CORT118335 are under investigation to restore glucocorticoid sensitivity. Mifepristone shows efficacy in treating psychotic depression and Cushing’s syndrome; its use in inflammatory diseases is limited by its antagonistic profile (McGinn et al., 2021). The development of GR modulators that selectively enhance anti-inflammatory transrepression while minimizing metabolic transactivation effects is an active area of research. Moreover, cytokine inhibitors, particularly those targeting interleukin-6 (IL-6) and tumor necrosis factor (TNF), have been evaluated in stress-related inflammatory and psychiatric conditions. For instance, tocilizumab (anti-IL-6R) and infliximab (anti-TNF) have shown mixed results in depression and post-traumatic stress disorder (PTSD), particularly in patients with elevated inflammatory markers (Mrđa et al., 2025). However, these biologics are costly, require parenteral administration, and may not be effective in individuals with low baseline inflammation. The future of cytokine inhibition can be transformed through biomarker-guided patient stratification, enabling more precise targeting of individuals likely to benefit from these therapies.
The discovery of the inflammatory reflex, a neural circuit involving the vagus nerve that regulates immune responses, has led to the development of neuromodulatory therapies aimed at reducing systemic inflammation through non-pharmacological means. Vagus nerve stimulation (VNS) is one of the most studied interventions in this domain. VNS inhibits cytokine production via the α7 nicotinic acetylcholine receptor (α7nAChR) by activating the cholinergic anti-inflammatory pathway (Singh et al., 2025). Clinical studies report benefits in rheumatoid arthritis and inflammatory bowel disease, indicating that modulation of this reflex can translate into measurable reductions in disease activity. VNS is generally safe and tolerable over long durations; however, device implantation, cost, and inter-individual variability in response limit scalability (Calderon-Martinez et al., 2024). To address these barriers, current work is evaluating non-invasive transcutaneous VNS (tVNS), which preserves the mechanistic rationale of cervical VNS while improving accessibility and risk profiles. Bioelectronic medicine builds on this concept by using implantable or wearable devices to precisely modulate neural circuits that regulate inflammation. The SetPoint Medical trial, for example, showed that vagus nerve stimulation significantly reduced disease activity in patients with rheumatoid arthritis (Peterson et al., 2024). Despite its promise, bioelectronic therapy faces challenges including device miniaturization, regulatory approval, and patient compliance. Additionally, Microglial inhibitors, such as minocycline, have also shown potential in modulating neuroinflammation, particularly in depression, PTSD, and neurodegenerative diseases. Minocycline exerts mild antidepressant effects in clinical trials, but its broad-spectrum antibiotic activity raises concerns about microbiome disruption and antimicrobial resistance (Abdul Shaik et al., 2025). More selective microglial modulators, such as TSPO ligands and P2X7 receptor antagonists, are currently under investigation as safer alternatives (Singh et al., 2025).
Psychosocial and behavioral interventions provide a low-risk, non-invasive means of reducing the physiological burden of chronic stress, with measurable effects on immune and metabolic function. Evidence supports cognitive-behavioral therapy (CBT), mindfulness-based stress reduction (MBSR), and related stress-management techniques in lowering systemic inflammatory markers (O’Toole et al., 2025). Across studies, these interventions are associated with reductions in C-reactive protein (CRP) and interleukin-6 (IL-6) and with improved immune function in individuals experiencing chronic stress, depression, or autoimmune conditions. Notably, MBSR has also been linked to increased telomerase activity, suggesting a potential effect on cellular aging. Their effectiveness, however, depends heavily on patient engagement and on access to trained professionals. Digital therapeutics and mobile health applications are expanding reach and enabling remote delivery, although large-scale clinical validation is still in progress (Mrđa et al., 2025). Furthermore, Lifestyle measures complement these approaches in addressing stress-related immunometabolic dysregulation. Regular physical activity enhances anti-inflammatory cytokine production (e.g., IL-10), improves insulin sensitivity, and promotes neuroplasticity. Dietary patterns such as the Mediterranean diet, characterized by higher intake of polyphenols, fiber, and omega-3 fatty acids, demonstrate anti-inflammatory and neuroprotective effects (Suárez-Cuenca et al., 2025). These strategies are cost-effective and scalable, but adherence remains a central limitation. Looking forward, personalized approaches informed by genetic, microbiome, and metabolic profiling can improve engagement and optimize outcomes by matching interventions to individual biological and behavioral profiles (Liang et al., 2023).
Stem-cell–based interventions, particularly those using mesenchymal stem cells (MSCs), have shown immunomodulatory and neuroprotective properties in preclinical models of stress-related disorders. MSCs reduce neuroinflammation, restore synaptic plasticity, and promote tissue repair (Han et al., 2025). Clinical trials in autoimmune conditions such as multiple sclerosis and graft-versus-host disease have demonstrated therapeutic potential, with preliminary evidence suggesting benefits in treatment-resistant depression. However, challenges such as cell sourcing, immune rejection, and long-term safety remain significant barriers to widespread clinical application. Additionally, epigenetic therapies offer a novel approach to addressing the molecular legacy of chronic stress. Persistent epigenetic changes, including DNA methylation and histone modifications, can alter gene expression in immune and neural cells, contributing to long-term vulnerability to stress-related disorders (Mallick & Duttaroy, 2023). Histone deacetylase (HDAC) inhibitors such as valproic acid and vorinostat, as well as miRNA modulators, are being investigated for their ability to reverse these changes. However, the broad effects of these agents on gene regulation raise concerns about off-target effects and potential tumorigenicity. Emerging technologies, such as CRISPR-dCas9-based epigenetic editing, offer the promise of more precise and targeted interventions in the future (Banushi et al., 2025).
Despite major advances in mapping the pathways of stress-induced neuroimmune dysfunction, significant gaps remain in translating these insights into effective and personalized therapies. Future research must therefore move beyond descriptive associations to integrated models that clarify causality, identify biomarkers, and develop interventions tailored to specific patient subgroups. Biomarker discovery is a key priority with conventional markers such as CRP and IL-6 provide useful but nonspecific signals. Newer approaches such as cerebrospinal fluid cytokine profiling, PET imaging of microglial activation, and epigenetic markers like NR3C1 and FKBP5 methylation offer more precise insights (Liakakis et al., 2025). Multi-omic platforms that combine genomics, transcriptomics, proteomics, and metabolomics hold particular promise for developing biomarker panels capable of supporting precision psychiatry (Mrđa et al., 2025).
Establishing causal mechanisms requires longitudinal studies that follow stress exposure, immune activity, and brain function over time, alongside experimental models that reflect chronic and developmental stress rather than acute exposures. These insights will support therapeutic innovation by identifying which patients benefit from targeted anti-inflammatory or epigenetic interventions. For example, TNF-α inhibitors may only be effective in patients with high inflammatory load, while HDAC inhibitors or BDNF-restoring agents may target those with specific epigenetic or neurotrophic deficits. Clinical trials should therefore adopt biomarker-based inclusion to improve precision and overcome past inconsistencies. Digital health technologies offer new opportunities for continuous, real-world monitoring. Wearables measuring heart rate variability, sweat cortisol, or salivary cytokines, combined with smartphone-based digital phenotyping, could track neuroimmune risk states and predict relapse (Jolly et al., 2025). These approaches could enable timely, adaptive interventions, although issues of validation, privacy, and algorithm transparency must be addressed before widespread use (Li et al., 2023). Finally, as neuroimmune biomarkers move closer to clinical application, ethical and policy considerations must guide implementation. While predictive tools can improve prevention and treatment, they also carry risks of stigmatization and unequal access. Close collaboration between researchers, clinicians, and policymakers will be necessary to ensure that advances are translated into safe, equitable, and ethically sound healthcare practices (Peterson et al., 2024).
Neuroimmune interactions provide the principal mechanism through which chronic stress influences health, linking dysregulation of the HPA axis, sustained pro-inflammatory cytokine activity, and glial activation to wide-ranging effects on brain function. Stress functions primarily as an amplifier of pre-existing biological susceptibilities shaped by genetics, hormones, the microbiome, and environmental exposures. It acts through prolonged neuroimmune activation, degrades synaptic plasticity, cognition, and emotional regulation. These factors thereby increase vulnerability to psychiatric disorders, neurodegeneration, and inflammatory conditions, and concurrently aggravate cardiometabolic and oncologic risk via persistent inflammatory and autonomic imbalance. Translational progress depends on treating this amplifier role explicitly in study design and care pathways. Emerging therapeutics include cytokine antagonists, glucocorticoid receptor modulators, microbiome-targeted interventions, and wearable biosensors for autonomic and inflammatory monitoring. Future research should focus on biomarker-guided, longitudinal, systems-level validation and advance a precision-medicine framework that integrates multi-omics profiling and digital health tools. This will enhance individualized risk stratification to identify stress-responsive endotypes and to time interventions to periods of highest biological leverage. A unified, transdisciplinary effort spanning neuroscience, immunology, endocrinology, psychiatry, and public health is essential to convert mechanistic insight into scalable strategies that improve neuroimmune health and clinical outcomes.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)