Keywords
Choroid plexus tumors, Choroid plexus papillomas, Choroid plexus carcinoma, Pediatric neurosurgery, Surgical outcomes, Histopathology, Hydrocephalus
Choroid plexus tumors, Choroid plexus papillomas, Choroid plexus carcinoma, Pediatric neurosurgery, Surgical outcomes, Histopathology, Hydrocephalus
Choroid plexus tumors (CPTs) are rare intraventricular neoplasms arising from the epithelial cells of the choroid plexus, a structure essential for cerebrospinal fluid (CSF) production and regulation.1,2 Their incidence varies with age, accounting for approximately 1–4% of pediatric brain tumors and 0.4–1% of intracranial tumors in adults.3
Because of their intraventricular location and frequent CSF overproduction, CPTs commonly cause hydrocephalus and intracranial hypertension, with clinical manifestations depending on tumor size and ventricular site.3,4 Diagnosis is primarily based on magnetic resonance imaging (MRI) and confirmed by histopathological examination.5 According to the 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System (5th edition), CPTs are classified as choroid plexus papilloma (CPP, WHO grade I), atypical choroid plexus papilloma (aCPP, WHO grade II), and choroid plexus carcinoma (CPC, WHO grade III), the latter representing an aggressive malignant entity.6,7
Surgical resection remains the cornerstone of treatment, although management is often challenging because of marked tumor vascularity and complex anatomy, particularly in infants and young children.8,9
Given the rarity of CPTs and the heterogeneity of their presentation and outcomes, we report our institutional experience, focusing on clinical, radiological, histopathological, therapeutic, and outcome-related characteristics.
We conducted a retrospective descriptive study including all patients treated for choroid plexus tumors at the Neurosurgery Department of Habib Bourguiba University Hospital (Sfax) between January 2010 and December 2023, regardless of age or sex.
Clinical data included age, sex, presenting symptoms, neurological examination findings, and diagnostic delay. Radiological evaluation was based on CT and MRI, assessing tumor location, laterality, ventricular site, parenchymal extension, hydrocephalus, calcifications, mass effect, and contrast enhancement.
Surgical variables included time to surgery, preoperative cerebrospinal fluid diversion, surgical approach, intraoperative tumor characteristics, extent of resection, operative duration, and perioperative complications. Histopathological analysis focused on tumor subtype, cellularity, nuclear pleomorphism, mitotic activity, necrosis, solid growth patterns, and immunohistochemical findings.
Postoperative outcomes, adjuvant treatments, complications, recurrence, and survival were recorded during follow-up at 3 months, 1 year, and 5 years.
Analysis was based on the calculation of medians with ranges for quantitative variables and frequencies for qualitative variables.
Between January 2010 and December 2023, seven patients with choroid plexus tumors were treated at our institution. Histologically, four tumors were choroid plexus papillomas (CPP) and three were choroid plexus carcinomas (CPC). Median age at diagnosis was 2 years (range: 2 months and 20 days–28 years), with six pediatric patients and one young adult. There was a female predominance (5 females, 2 males).
The median diagnostic delay was 7 days (range: 1 day–2 months). The most frequent presenting symptoms were vomiting (n = 6) and headache (n = 3). Visual disturbances were observed in three patients, seizures in two, and focal neurological deficits or altered consciousness in one patient each.
On neurological examination, macrocephaly and sunset gaze were each noted in one infant. Motor deficits were present in two patients, cerebellar signs in one, sixth cranial nerve palsy in one, and papilledema in one. Café-au-lait spots were observed in one patient. Neurological examination was otherwise normal in two cases ( Table 1).
| Obs | Age/Sex | Clinical features | Imaging summary |
|---|---|---|---|
| 1 (CPP) | 28 F | Occipital headache, nausea, diplopia; cerebellar signs, CN V & VI deficits | 4th ventricle to CPA, 6×6 cm lesion, cystic + solid, heterogeneous enhancement, triventricular hydrocephalus (Figure 1) |
| 2 (CPP) | 2.8 F | Holocranial headache, projectile vomiting; clinically normal | Left occipital horn of the lateral ventricle, 5×4.7×3.8 cm, isodense, homogeneous enhancement, quadriventricular hydrocephalus (Figure 2) |
| 3 (CPP) | 1.75 F | Vomiting, altered consciousness (GCS 7/15) | Lateral extented to 3rd ventricle 3.7×2.7×2.4 cm, lobulated, hyperdense; cauliflower-like lesion, intense homogeneous enhancement |
| 4 (CPP) | 0.17 F | Projectile vomiting, feeding refusal, macrocephaly; hypotonia, sunset gaze | 6×4×2.5 cm intraventricular, irregular contours, microcalcifications, intense post-contrast enhancement, quadriventricular hydrocephalus |
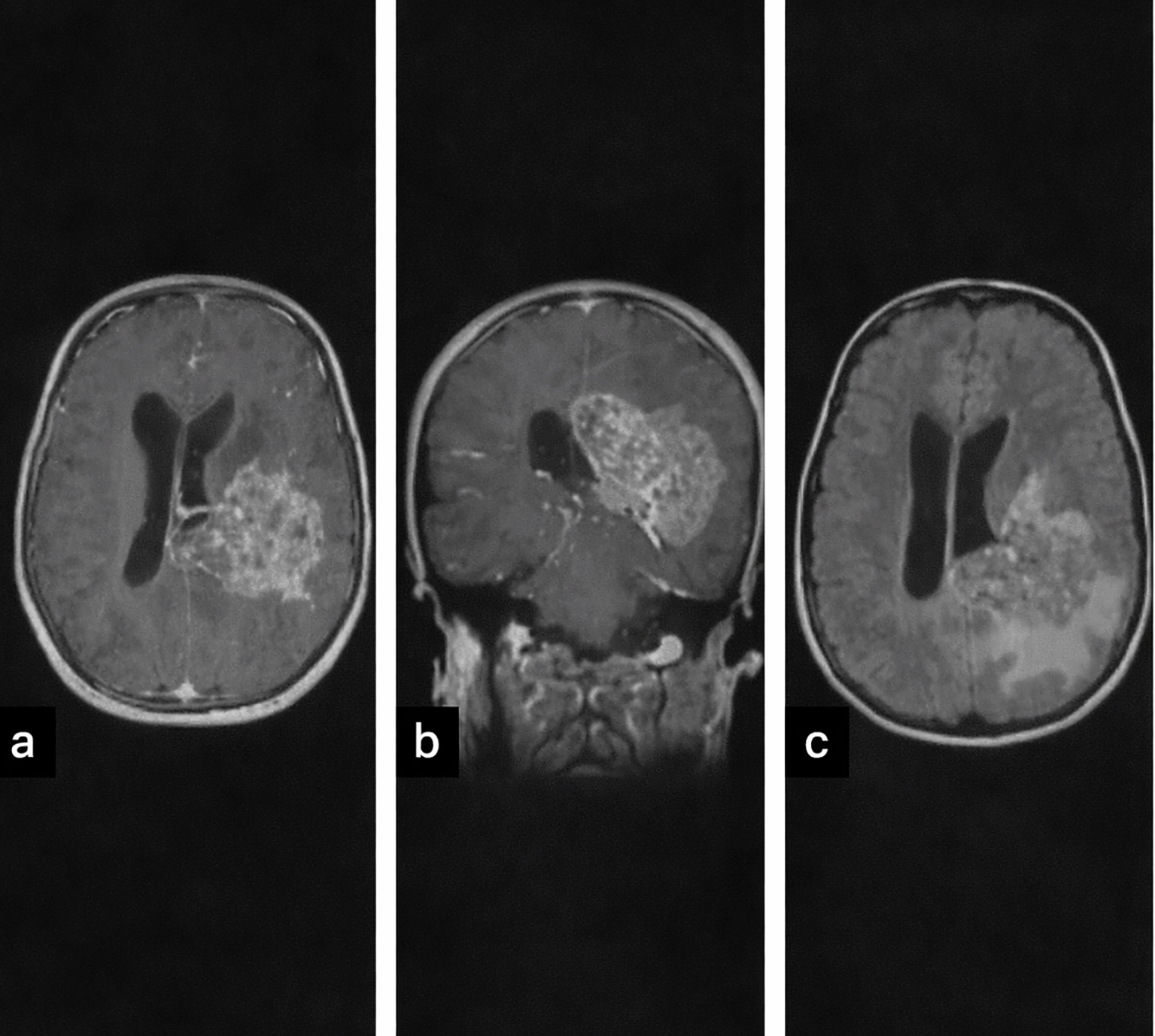
| 5 (CPC) | 9 F | Generalized tonic-clonic seizure; right hemiparesis, papilledema | Heterogeneous, hyperdense, calcifications, hemorrhage, perilesional edema; heterogeneous enhancement, parenchymal invasion (Figure 3) |
| 6 (CPC) | 3 M | Headache, vomiting, seizures; clinically normal | 11 cm solid lesion, heterogeneous enhancement, calcifications, marked mass effect |
| 7 (CPC) | 1.75 M | Left-sided weakness, vomiting; left hemiparesis | V3 origin, 8×6 cm hyperdense lesion, hemorrhagic foci, perilesional edema; heterogeneous signal, strong enhancement, biventricular hydrocephalus |
Tumors were located in the lateral ventricles in five cases (Figure 1), the third ventricle in one case, and the fourth ventricle with extension to the cerebellopontine angle (CPA) in one case (Figure 2). CT imaging (performed in six patients) showed lobulated intraventricular masses that were iso- to hyperdense with homogeneous enhancement in CPPs, frequently associated with hydrocephalus. CPCs appeared heterogeneously hyperdense with intense enhancement and intratumoral calcifications in two of three cases.
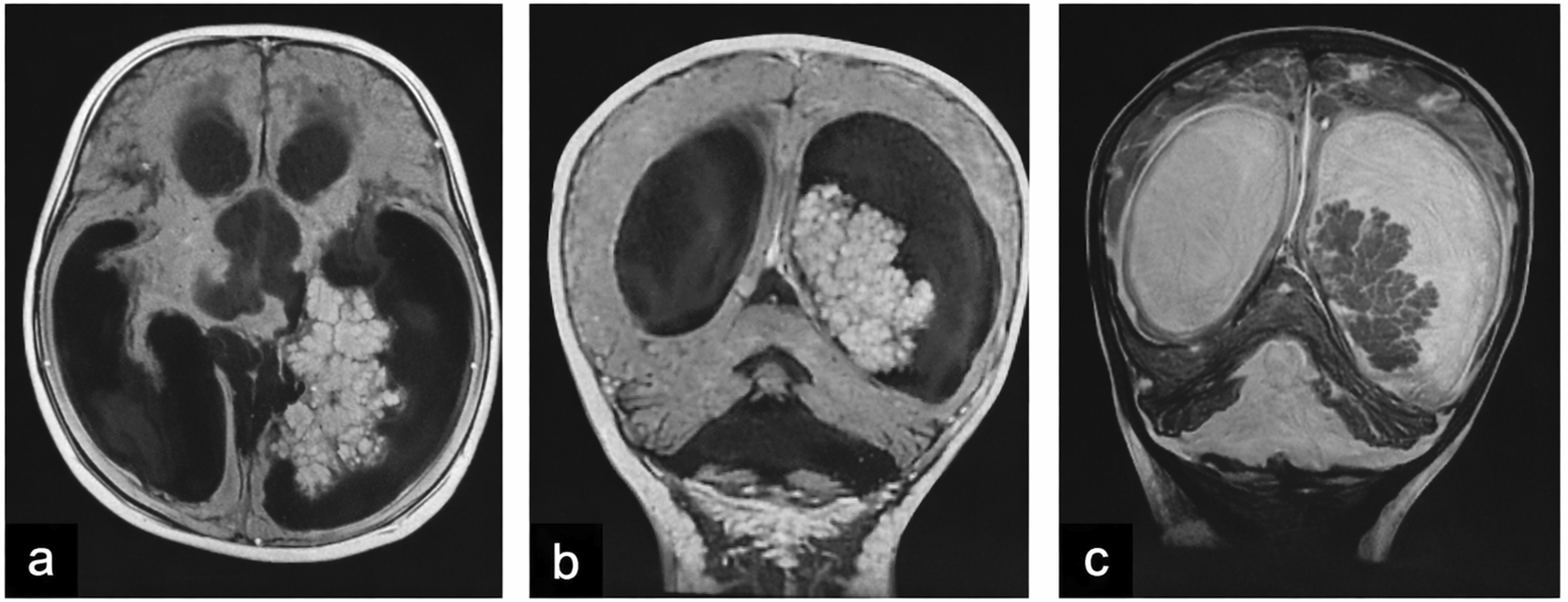
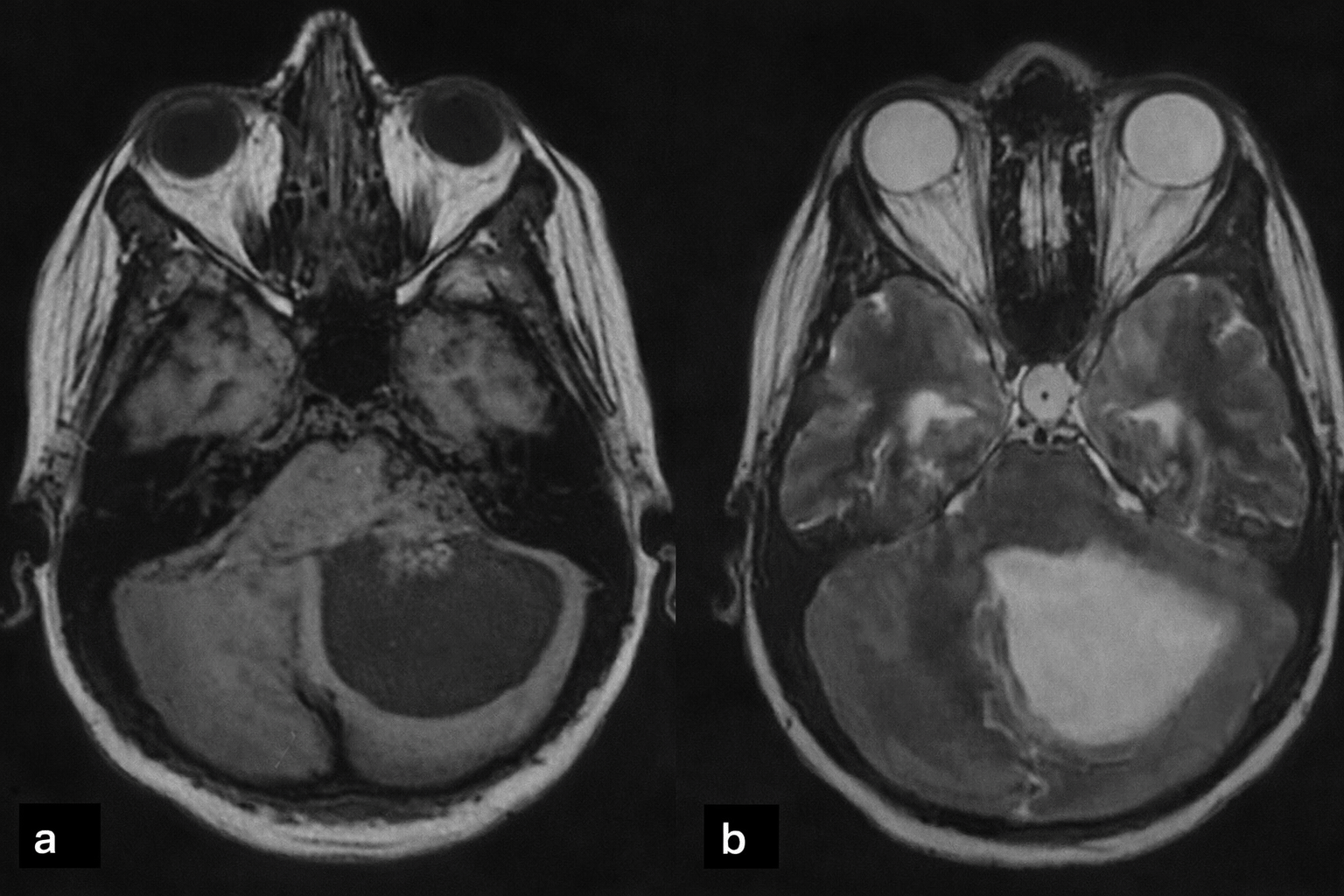
MRI (performed in five patients) demonstrated variable signal characteristics. CPPs were typically well-circumscribed and intensely enhancing, while CPCs appeared heterogeneous (Figure 3), with areas of calcification, hemorrhage, and parenchymal invasion. Hydrocephalus was present in the majority of cases ( Table 1).
All patients underwent surgical intervention. Gross total resection was achieved in four patients, near-total resection in one, incomplete resection in one, and large biopsy in one. CPPs were generally friable and minimally hemorrhagic, whereas CPCs were highly vascular and hemorrhagic. Uncontrollable intraoperative bleeding occurred in one patient.
Preoperative external ventricular drainage was required in two patients. Postoperatively, permanent cerebrospinal fluid diversion was necessary in two cases: one ventriculoperitoneal shunt following failed EVD weaning and one subdural-peritoneal shunt for postoperative subdural hygroma.
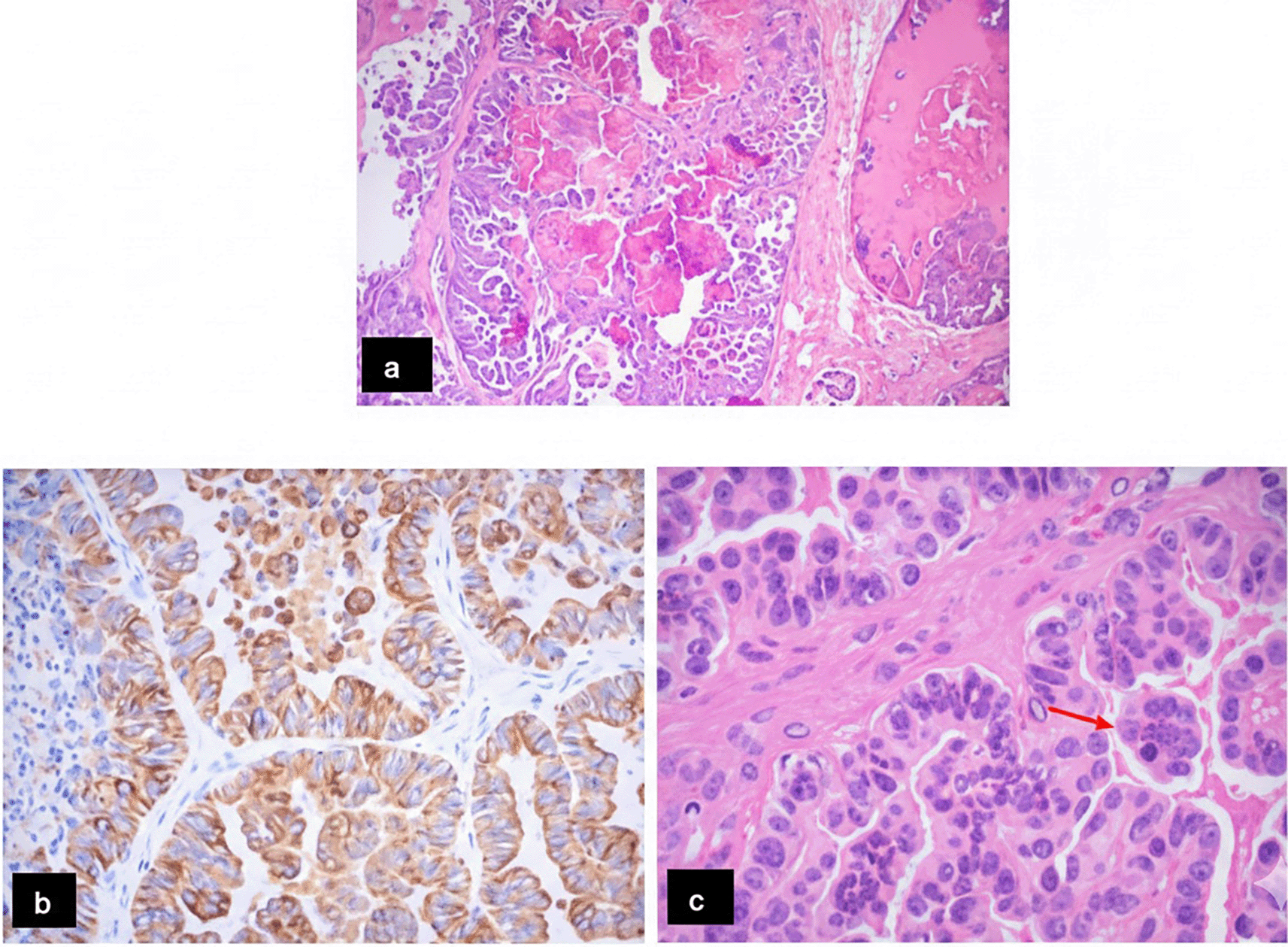
CPPs demonstrated typical papillary architecture with thin fibrovascular cores lined by monomorphic cuboidal epithelial cells, low cellularity, absence of necrosis, and no mitotic activity. CPCs exhibited high cellularity, nuclear pleomorphism, solid growth areas, frequent mitoses, and necrosis in two cases. Immunohistochemistry confirmed epithelial differentiation in all CPCs, with a Ki-67 index up to 40% in one case (Figure 4 and Table 2).
All patients with CPP had a favorable postoperative course, with no tumor recurrence during follow-up ranging from 3 to 13 years. Two CPP patients required shunting for postoperative hydrocephalus or subdural hygroma.
Among CPC patients, outcomes were poor. One patient died intraoperatively from hemorrhagic shock, another died on postoperative day 4 from myocardial infarction, and the third developed postoperative meningitis with residual hemiparesis and died shortly thereafter before initiation of chemotherapy ( Table 3).
Choroid plexus tumors (CPTs) are rare intraventricular neoplasms, predominantly affecting the pediatric population, particularly during the first years of life.1,3 In our series, the median age at diagnosis was 2 years, with six of seven patients being children, reflecting the well-documented pediatric predominance of these tumors. The sex ratio was 2:5, indicating a slight female predominance, contrasting with some reports of equal distribution or slight male predominance.8 Tumor characteristics varied by sex, location, and histology: cerebellopontine angle tumors were associated with older age, benign histology, and female sex,4,10 consistent with our oldest patient (28 y, CPP, 4th ventricle extending to CPA). In contrast, infantile CPTs were mostly lateral ventricular and more common in males.1
Clinical presentation was dominated by symptoms of increased intracranial pressure, mainly vomiting and headache, reflecting the high prevalence of hydrocephalus in CPTs. The median diagnostic delay was short (7 days), likely related to the rapid onset of symptoms in pediatric patients, particularly infants, in whom hydrocephalus manifests early with macrocephaly, lethargy, or irritability. This contrasts with adult patients, in whom intraventricular tumors may remain asymptomatic for longer periods.9,11,12
Radiologically, our findings were concordant with the literature. CPPs typically appeared as well-circumscribed intraventricular masses with homogeneous enhancement, whereas CPCs demonstrated heterogeneous enhancement, calcifications, and parenchymal invasion causing vasogenic edema.12,13 Diffusion-weighted imaging shows hypercellularity with restricted diffusion, and spectroscopy may help distinguish CPPs (myo-inositol peak) from CPCs (choline elevation).11,14 Despite these suggestive features, differentiation between CPP and CPC based solely on imaging remained difficult, reinforcing the central role of histopathological examination for definitive diagnosis.
Surgical management remains the cornerstone of treatment.8,15 Gross total resection was achieved in the majority of CPP cases and was associated with excellent long-term outcomes, with no recurrences observed during follow-up. Management of CPCs remains challenging: complete excision is the goal but often difficult due to hypervascularity and invasive behavior. Infants and young children are particularly vulnerable to blood loss; excessive intraoperative bleeding can result in perioperative mortality,16 as observed in one of our patients. Collaboration with anesthesiology teams experienced in pediatric neuro-oncology is essential. Preoperative embolization has been proposed to reduce bleeding but is technically challenging due to small, tortuous feeding vessels17; MR angiography can assist in delineating the surgical corridor.
CSF diversion is also a critical component of management.18 Hydrocephalus, common in CPTs, may rapidly impair consciousness and sometimes requires urgent external ventricular drainage (EVD). Hydrocephalus often resolves after tumor resection, allowing EVD removal, but persistent obstruction due to necrotic debris, hemorrhage, or arachnoiditis may necessitate ventriculoperitoneal shunting.4,8 Thus careful monitoring of ventricular size and intracranial pressure is mandatory. Postoperative subdural hygroma, a recognized complication after transcortical approaches in children, was also observed and successfully managed with cerebrospinal fluid diversion.18
Histopathological examination remains the gold standard for the definitive diagnosis and grading of choroid plexus tumors. In our series, histological features were fully consistent with the 2021 WHO Classification of Tumors of the Central Nervous System (5th edition). CPPs displayed classic papillary architecture with low cellularity, absent atypia, and negligible mitotic activity, consistent with indolent, low-grade behavior, whereas CPCs exhibited high cellularity, nuclear pleomorphism, necrosis, and elevated Ki-67 (up to 40%), reflecting aggressive malignancy in line with WHO criteria. The diagnosis of CPC requires the presence of at least four of five histological features—high cellularity, nuclear pleomorphism, loss of papillary architecture, necrosis, and increased mitotic activity—which were met in our CPC cases.6,7,19 Immunohistochemistry confirmed the epithelial origin of CPCs, which showed high Ki-67 proliferation indices (up to 40%) reflecting aggressive behavior, whereas CPPs exhibited very low Ki-67 (<1%), consistent with indolent biology.12,19
At the molecular level, alterations of the TP53 pathway play a central role in choroid plexus tumorigenesis, particularly in choroid plexus carcinomas, where TP53 mutations are reported in up to 50% of cases.20,21 Germline TP53 mutations, as seen in Li–Fraumeni syndrome, are strongly associated with pediatric CPC and carry important prognostic and therapeutic implications.5,12,22
For CPCs, chemotherapy after incomplete resection can reduce tumor size and vascularity. From a therapeutic perspective, adjuvant chemotherapy and radiotherapy have been shown to improve survival in CPC patients, particularly following incomplete resection.16,23 However, the use of radiotherapy in infants is limited by its long-term neurocognitive and endocrine toxicity.24 In our series, adjuvant treatment could not be administered due to early postoperative mortality or rapid clinical deterioration, which likely contributed to the poor outcomes observed in CPC patients.
Successful management of pediatric CPTs requires a multidisciplinary approach, meticulous preoperative planning, microsurgical expertise, proactive CSF management, and close postoperative surveillance. Tailoring the surgical strategy to tumor type, patient age, and vascular characteristics significantly reduces perioperative morbidity and improves long-term outcomes.
Overall, our findings reinforce the heterogeneity of CPTs. Complete surgical resection remains the key determinant of outcome in CPTs, while CPCs continue to carry a poor prognosis, highlighting the need for improved perioperative strategies and molecular-guided therapies.
The main limitation of this study is the small sample size inherent to the rarity of CPTs, as well as its retrospective nature. Molecular analyses, particularly TP53 status, were not available, limiting biological stratification. Nevertheless, this series provides a detailed clinico-radiological and pathological correlation with long-term follow-up, illustrating real-life surgical challenges and outcomes.
This retrospective descriptive study was conducted in accordance with the principles of the Declaration of Helsinki. Authorization to access patient medical records was obtained from the administration of Habib Bourguiba University Hospital. The study involved the analysis of anonymized data collected as part of routine clinical care, with no additional diagnostic or therapeutic procedures. No patient-identifying information was included. Given the retrospective, non-interventional nature of the study, formal approval from a local Institutional Review Board or Ethics Committee was not required, and the requirement for informed consent was waived in accordance with institutional policies.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)