Keywords
statistical resampling, RAWR, phylogenetic support estimation, phylogenetic tree, multiple sequence alignment, phylogenetics, GUI software, API
This article is included in the Evolutionary Genomics collection.
statistical resampling, RAWR, phylogenetic support estimation, phylogenetic tree, multiple sequence alignment, phylogenetics, GUI software, API
Statistical resampling has become an essential technique that is used in a wide variety of biomolecular sequence analysis tasks. The standard bootstrap method (Efron, 1979) is among the most widely used general-purpose resampling methods. A particularly important application of bootstrap resampling is confidence interval estimation for phylogenetic reconstruction (also known as phylogenetic support estimation) (Felsenstein, 1985). The original paper describing this application is the 41st most cited in history (Van Noorden et al., 2014).
However, the bootstrap method make a crucial assumption that all sites in the input sequences are independent and identically distributed (i.i.d.). This assumption is an over-simplification for biomolecular sequence data due to evolutionary processes such as sequence insertions, deletions, and genetic recombination, as well as biomolecular structure and other factors – all of which can cause site dependencies within sequences.
A sequence-aware statistical resampling method known as RAWR (“RAndom Walk Resampling”) (Wang et al., 2021) was introduced to relax this simplifying assumption. Using either unaligned or aligned sequence data as input, RAWR resamples data by conducting a random walk directly on the input sequences. RAWR-resampled sequences satisfy a “neighbor preservation” property: neighboring bases in a resampled sequence are guaranteed to also appear as neighbors in the original input’s corresponding sequence. The original study of RAWR focused on the application of statistical resampling to phylogenetic tree support estimation. On simulated and empirical benchmarks with a range of dataset sizes and evolutionary divergence, RAWR yielded comparable or superior type I and type II error versus the phylogenetic bootstrap and state-of-the-art methods such as aLRT (Anisimova and Gascuel, 2006) and TBE (Lemoine et al., 2018).
In this manuscript, we present the RAWR v1.0 software package for sequence-aware statistical resampling of biomolecular sequence data. The release includes applications to two essential tasks: support estimation for multiple sequence alignment and phylogenetic tree reconstruction. The software distribution includes user-friendly clients and developer-friendly software libraries. Highlights of the package include phylogeny visualization, asynchronous email delivery of results, parallel processing, and an Application Programming Interface (API) to integrate RAWR resampling into third-party software distributions in bioinformatics and beyond.
The RAWR version 1.0 software distribution enables sequence-aware statistical resampling of biomolecular sequence data. Resampling is performed using the RAWR technique, which resamples sites during a random walk along biological molecular sequences while preserving site neighbor relationships. Two main resampling tasks are supported: (1) phylogenetic tree support estimation and (2) MSA support estimation. The phylogenetic tree support estimation task is defined as follows: the input is a phylogenetic tree estimate on a set of unaligned sequence data, and the output consists of a set of support values. Each support value annotates a distinct edge in the input tree estimate based on the proportion of re-estimated trees that also display that edge, where re-estimation is performed on RAWR-resampled replicates. The MSA estimation task is similar, except that the annotation object and re-estimates are MSAs (rather than phylogenetic trees) and support values annotate nucleotide/residue pair homologies in an annotation MSA (rather than edges in an annotation tree).
RAWR v1.0 contains three different Python implementations for end users: (1) a GUI desktop/PC client, (2) a local Galaxy client, and (3) a local web server. Detailed instructions for installing and using the three implementations are available in the Supplementary Appendix.
In addition to the end user implementations, an application programming interface (API) is provided for developer access to software distribution functions. The API can be used to integrate RAWR resampling and support estimation into third-party software and software pipelines.
GUI Desktop client. The GUI client provides a user-friendly window-based layout. Users interact with the GUI using drop down selection menus to configure analysis settings, buttons to choose file inputs, and a checkbox to enable parallelization ( Figure 1). The GUI desktop client is available for Windows, macOS, and Linux operating systems. Phylogeny visualization makes use of the ETE3 software library (Huerta-Cepas et al., 2016), and MSA support visualization is supported by JalView (Waterhouse et al., 2009). Multiprocessing is supported on computing architectures that are compatible with parallelization.

The client provides a user-friendly window-based interface for RAWR resampling analyses. Two primary resampling tasks are supported: phylogenetic tree support estimation and MSA support estimation. The main window provides user controls for resampling analysis parameters as follows. “Input 1” accepts a FASTA-formatted MSA. “Input 2” accepts a Newick-formatted phylogenetic tree. The first optional parameter determines the resampling task, where “tree” denotes phylogenetic tree support estimation and “msa” denotes MSA support estimation. The second optional parameter determines the resampling algorithm, where “rawr” denotes the RAWR algorithm and “seres” denotes the SERES algorithm. The third and fourth parameters describe the number of resampled replicates (“Sample Number”) and random walk reversal rate (“Reverse Rate”), respectively. Finally, the fifth and sixth parameters are reserved for the SERES algorithm only and describe number and length of anchor regions to place in the MSA (“Anchor Number” and “Anchor Length”, respectively). Parallelization (“Multiprocess”) is optionally available to speed up analyses using a user-specified number of computing cores, and is specified using a drop-down menu. Default values for parameters are configured as shown.
As noted above, two primary resampling tasks are supported. In the phylogenetic tree support estimation task, the output is a Newick-formatted tree with phylogenetic support values. As an example, Figure 2 shows a phylogenetic tree with RAWR support values as visualized in the desktop client GUI. In the MSA support estimation task, the output is a text file annotating the original MSA with confidence intervals on nucleotide-nucleotide/residue-residue homologies as well as a JalView-compatible (Waterhouse et al., 2009) annotation file. Figure 3 provides an example visualization of the latter.
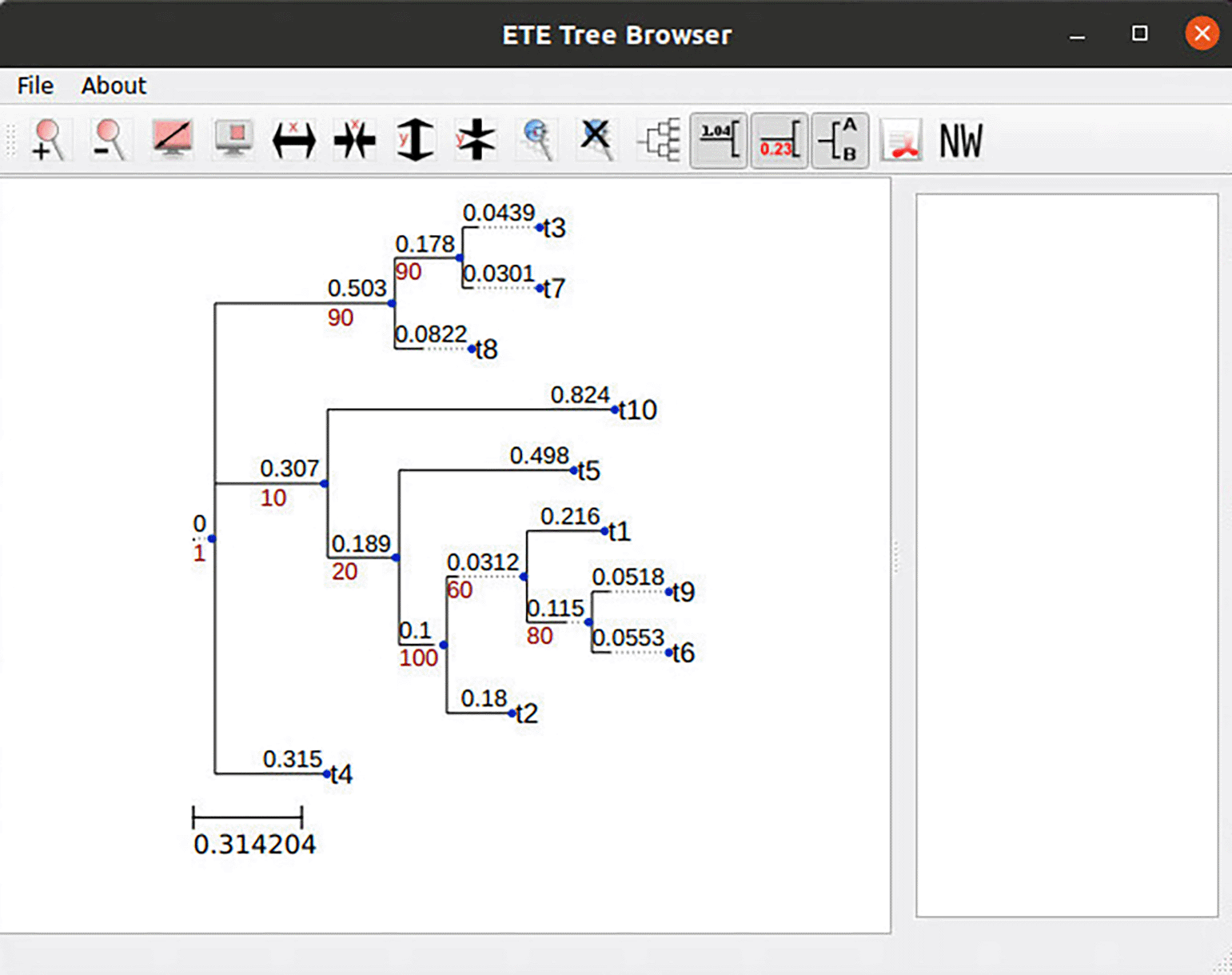
RAWR resampling was used to perform phylogenetic tree support estimation. The client utilizes the ETE (Huerta-Cepas et al., 2016) software package to perform the final tree visualization.
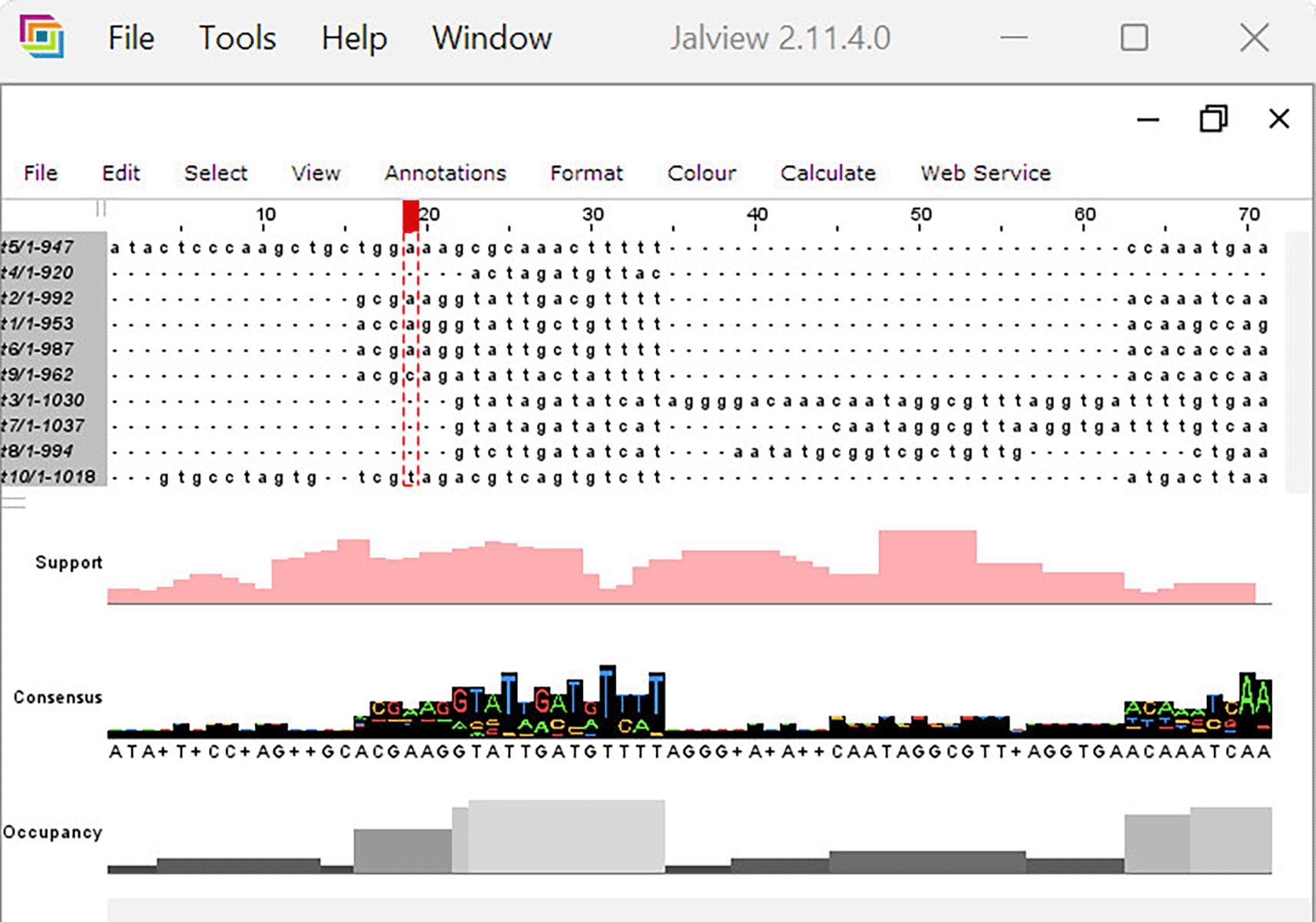
The client utilizes the JalView v2.11.4.0 (Waterhouse et al., 2009) software package to display the annotation MSA and RAWR support values (“Support” row).
Local Galaxy client. Galaxy (Giardine et al., 2005) is a popular open-source bioinformatics platform for both novice and experienced end users. The platform is developed for Linux and macOS operating systems. Many widely used bioinformatics algorithms, data processing procedures, and scientific workflows are included.
RAWR v1.0 can be served in a local Galaxy instance ( Figure 4). Visualization of RAWR analyses is enabled using Phylocanvas.gl (Abudahab et al., 2021). In addition to open source code, the RAWR software distribution includes binary executables for Debian GNU/Linux and macOS that were compiled using Python 3.10.
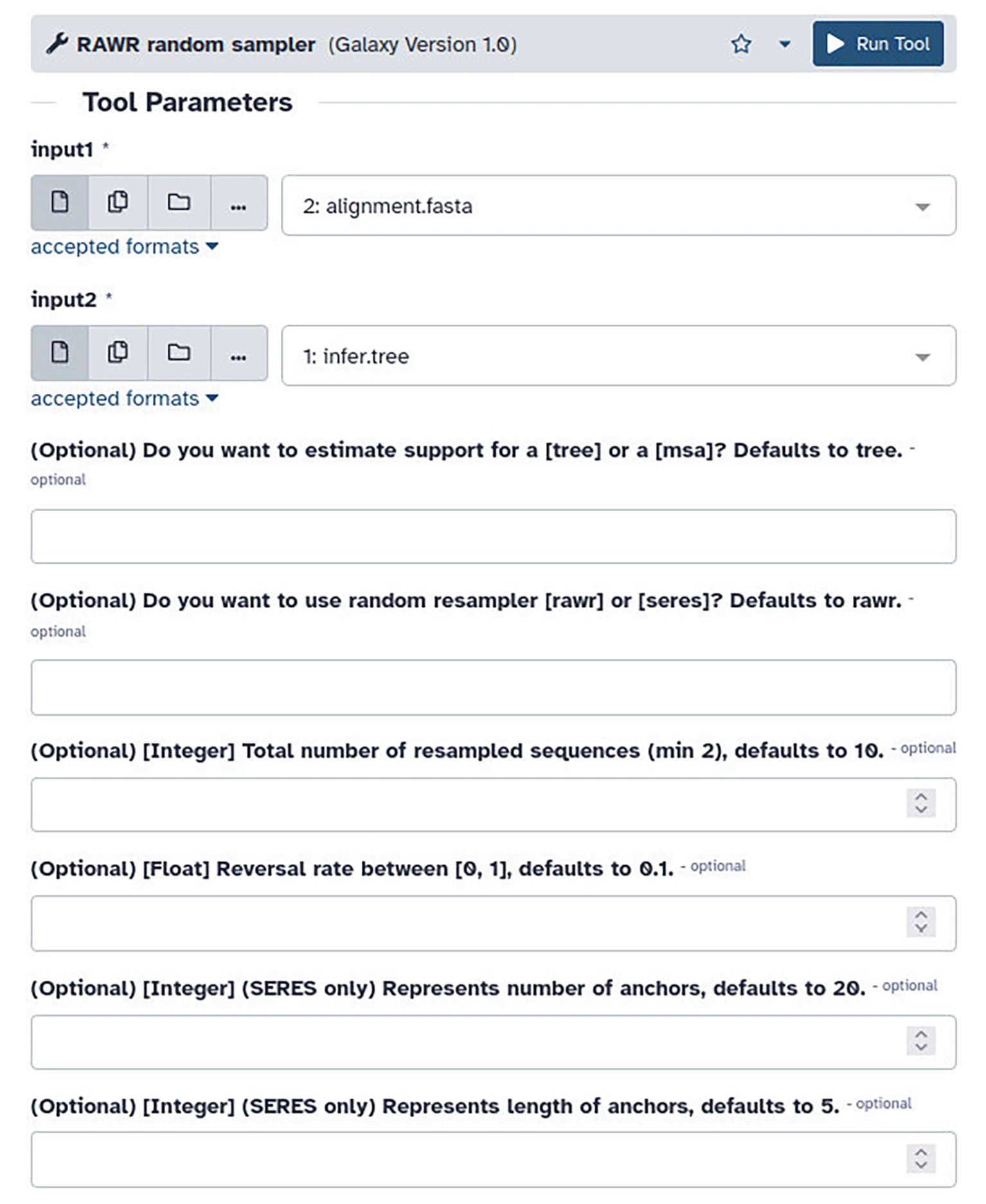
Parameter descriptions follow that of Figure 1.
Web client. The web client features a web form-based user interface with drop-down menus for selecting random walk resampling algorithms, radial buttons to open file inputs, and text boxes for parameters ( Figure 5). The web server software can be hosted on a macOS or Linux server. The web application offers an asynchronous processing option: submitted jobs are processed offline, the user provides an e-mail address to receive a notification email when the results are ready, and the email includes results as file attachments. Phylogenetic visualization support is enabled using the ETE3 software library (Huerta-Cepas et al., 2016).
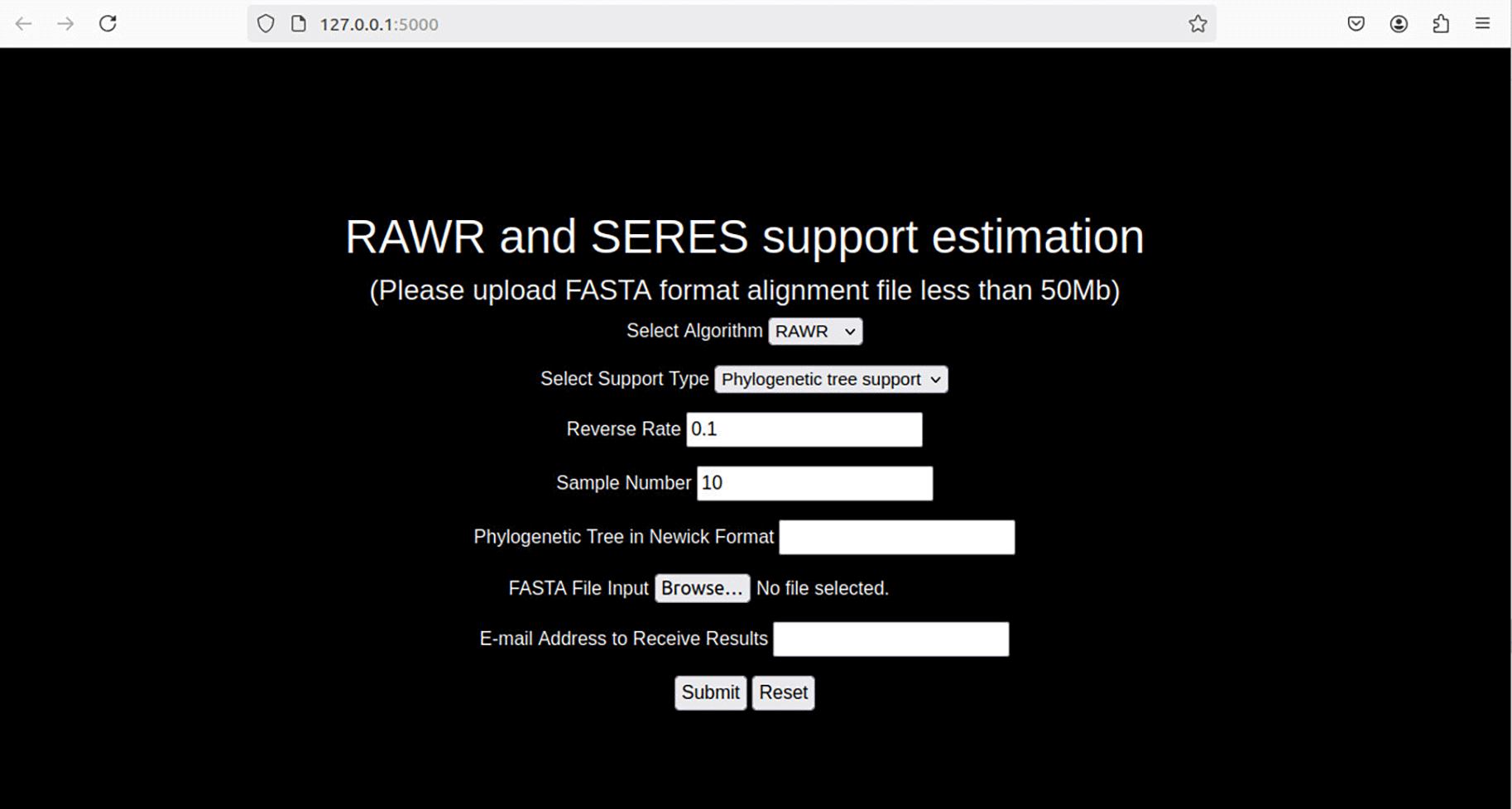
RAWR method parameters are user configurable in the web client. The “Select Algorithm” form field allows selection of RAWR resampling or SERES resampling (Wang et al., 2020), an alternative resampling approach that is related to and superseded by RAWR. The “Select Support Type” form field configures the resampling task to be either phylogenetic or MSA support estimation task. RAWR method parameters consist of a random walk reversal probability and the number of resampled replicates, and both can be configured (“Reverse Rate” and “Sample Number”, respectively). The input annotation tree and MSA are uploaded using the “Phylogenetic Tree in Newick Format” and “FASTA File Input” form fields respectively, where the former is specified in Newick format and the latter is specified in FASTA format. Asynchronous execution is enabled by providing an email address in the “E-mail Address to Receive Results” form field, and results sent via email when ready. (Some user interface elements are dynamically configured depending on the choice of resampling algorithm.)
Application programming interface (API). Developer access to and third-party software integration with the RAWR v1.0 software suite is enabled by an application programming interface (API). The RAWR API and software libraries are implemented in Python3. A Unified Modeling Language (UML) class diagram is shown in Figure 6. Example code is provided at https://github.com/kjl-msu/RAWR-web-software/blob/main/RAWR-tutorial.md.
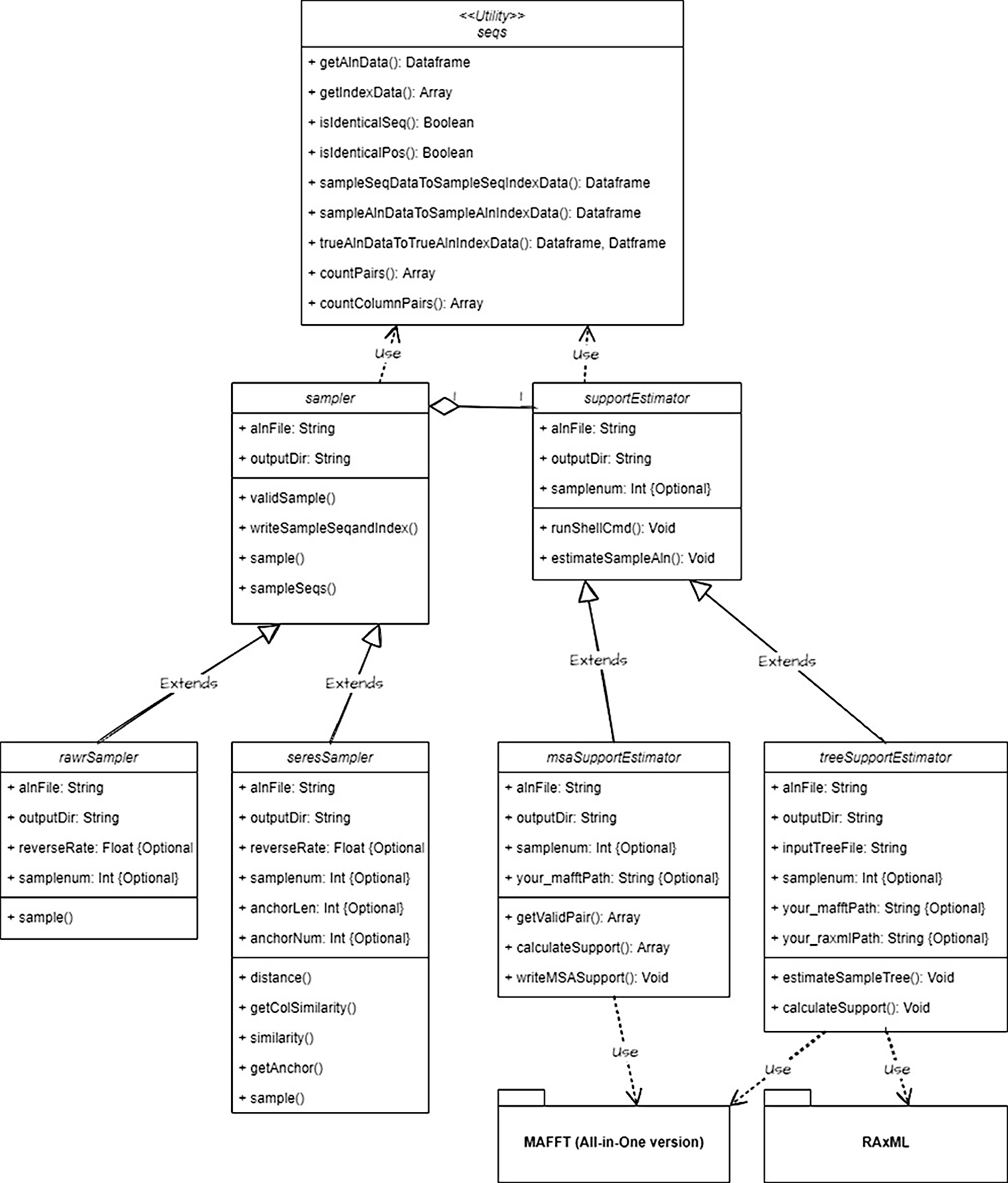
The RAWR v1.0 software suite includes a local Galaxy client, GUI desktop client, web server, and API. The minimum hardware requirements for the GUI desktop client and web server consist of a modern CPU with a minimum of 1 GB of RAM and file system storage of at least 150 MB. The minimum hardware requirements for the local Galaxy client are a modern CPU with a minimum of 4 GB of RAM and file system storage of at least 12 GB.
The RAWR v1.0 software suite has been developed for and tested on several major operating systems, with minimum software requirements as follows. The software suite is implemented in Python, with a minimum recommended version of 3.8 or newer. Python 3.8 is available on macOS version 10.9 or later, Windows 7 or later, and most modern Linux distributions (such as Ubuntu, CentOS, and Fedora). RAWR v1.0 has been tested in Python 3.8 and Python 3.10 using Virtualenv v20.26.6, and Anaconda v24.5.0 was used to manage Python package dependencies. The GUI desktop client and API are supported on macOS, Windows, and Linux operating systems. The desktop client has been tested on macOS Sequoia 15.0, Ubuntu 22.04 LTS, and Windows 11. The Galaxy and web server software are supported on macOS and Linux operating systems, and have been tested on macOS Sequoia 15.0 and Ubuntu 22.04 LTS. The web application has been tested on major web browsers, including Mozilla Firefox version 131 and Google Chrome version 129. Windows support for Galaxy or web server instances is enabled via virtualization or other emulation software (e.g., Windows Subsystem for Linux, Microsoft Hyper-V, etc.).
In this section, we walk through example use cases of the RAWR v1.0 software suite. The examples focus on two resampling tasks: phylogenetic tree support estimation and MSA support estimation. The walkthroughs utilize a simulated 10-taxon dataset from the performance study of Wang et al. (2021), and the dataset files are provided in the RAWR v1.0 software distribution under the rawr-software/example/dataset-10taxa/subdirectory.
Note that, as a prerequisite for the walkthroughs, the RAWR v1.0 software suite must be installed first. See Supplementary Online Materials for detailed software installation instructions.
Input files. An input MSA in FASTA format and an input annotation phylogeny in Newick format must be specified for RAWR analysis. The example dataset has these two inputs saved as the “alignment.fasta” file “infer.tree” file, respectively.
Output files. The RAWR v1.0 GUI desktop client and web server provides results in the following output files. A “samples” subdirectory contains the following files for each RAWR-resampled replicate: a metadata file that lists resampled column numbers in the input MSA with ordering based on RAWR resampling order, the corresponding resampled MSA, and the final set of resampled unaligned sequences that was resampled. The result of phylogenetic support estimation is the input annotation phylogeny with RAWR support values annotating branches; the output is provided in machine-readable Newick format as well as an image in the “tree.support.txt” and “tree.support.png” files, respectively. The result of MSA support estimation are provided in the “MSA.support.csv” output file. The output file is a comma-delimited spreadsheet that contains the following columns: (1) the position of a residue pair in the input MSA, (2) the row index of residue one in the pair, (3) the row index of residue two in the pair, and (4) a RAWR support value for the residue pair that is in the range [0, 1]. An accompanying feature annotation file “MSA.support.csv jalview annotation.txt” is generated alongside the MSA support output file.
The RAWR v1.0 local Galaxy client provides outputs for phylogenetic support estimation in the following output files. The Newick-formatted input tree with annotated support values is contained in the “tree.support.txt” output file. The result of MSA support estimation is a feature annotation file “MSA.support.csv jalview annotation.txt”, which can be visualized in JalView (Waterhouse et al., 2009).
GUI desktop client. We begin with a walkthrough for the GUI desktop client. A step-by-step description is provided for phylogenetic tree support estimation.
1. Assuming that the necessary Python dependencies have been installed in Anaconda, the conda environment is activated and the GUI client is then started.
$ cd rawr-software
$ conda activate python3.10
$ python main.py
2. The main window of the desktop client GUI appears ( Figure 7). Import input files using the “Select File” buttons. An example dataset is included in the RAWR v1.0 software distribution: to provide the dataset as input, select the “alignment.fasta” file for the “Input MSA” option and select the “infer.tree” file for the “Input Tree” option. Then, navigate to and/or create a new output folder by using the “Select Folder” button. We recommend that a new folder should be created for each RAWR analysis.
3. Optionally, edit the algorithm parameters on the lefthand side of the GUI client. Multiprocessing can also be enabled by checking the “Multiprocess” option. Default values for optional parameters are preset.
4. Run the analysis by selecting the “OK” button ( Figure 8). The progress bar will indicate the approximate percentage complete for the ongoing RAWR analysis.
5. Phylogenetic support estimation results are displayed in a new window ( Figure 2).
6. The output folder includes a “samples” subdirectory which contains RAWR-resampled replicates. If phylogenetic tree support estimation was the analysis task, a Newick file “tree.support.txt” and an image file “tree.support.png” will be located in the output folder. If MSA support estimation was selected, a MSA support result file “MSA.support.csv” and a feature annotation file “MSA.support.csv” jalview annotation will be located in the output folder.
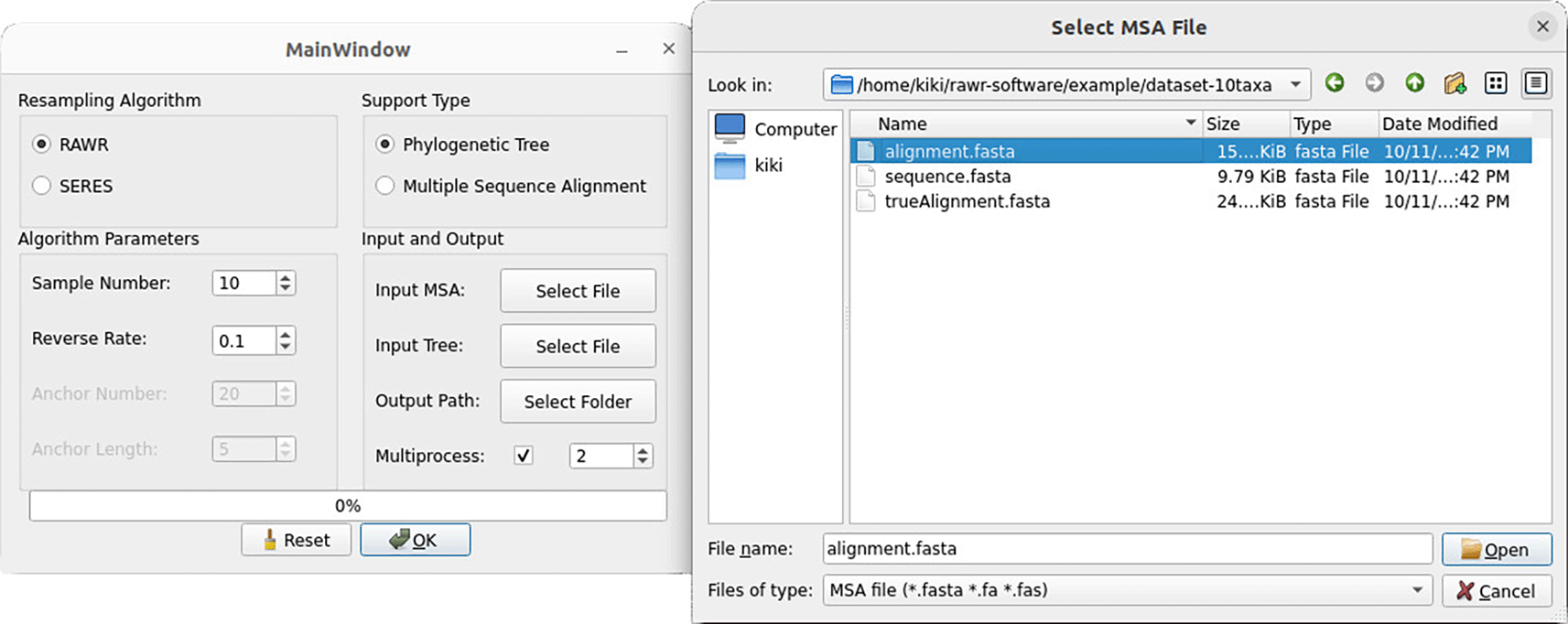
After clicking the “Select File” button next to the “Input MSA” option, a window dialog appears to select an input MSA file. The RAWR v1.0 software suite includes an example dataset which has an input MSA saved as the “alignment.fasta” file.
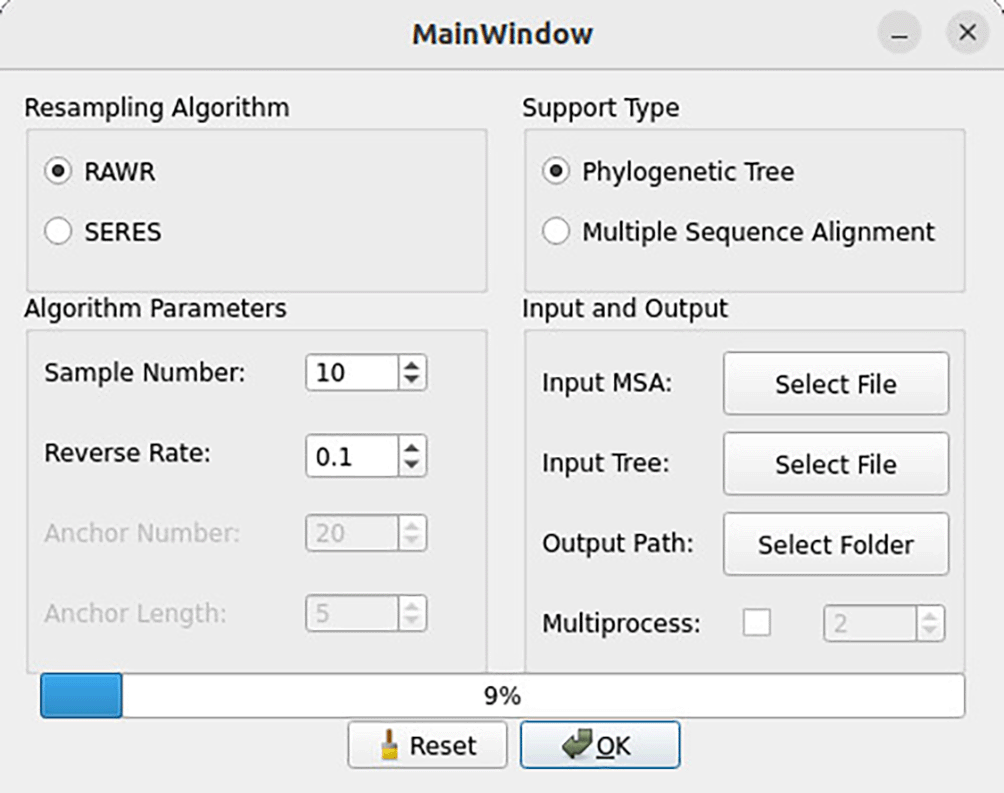
After selecting input files and method parameters using the main window, click the “OK” button to begin the RAWR analysis. A progress bar appears and reports the approximate percentage of the analysis that has completed.
Local Galaxy client. The following walkthrough uses the local Galaxy client to perform phylogenetic tree support estimation. The input files used for the other walkthroughs can also be used for the this walkthrough.
1. Assuming that local Galaxy client has been set up in Virtualenv, we activate the Virtualenv environment and use Planemo to serve a Galaxy instance.
$cd rawr-galaxy $. galaxy/.venv/bin/activate $planemo serve --galaxy_root galaxy
2. Import MSA and phylogeny files with the “Upload File” button on the left side of the GUI. Select file type “fasta” for the input MSA file and “newick” for the input tree file. Click “Start” to upload input files. The GUI dynamically updates as the upload proceeds ( Figure 9).
3. Navigate to “RAWR random sampler”, and the uploaded files will be automatically configured in the client window ( Figure 4). The default analysis configuration consists of a RAWR analysis to estimate phylogenetic tree support. Optional parameters are also prepopulated with default values (i.e., RAWR resampling is conducted with reversal probability γ = 0.1 to obtain a total of 10 RAWR replicates).
4. Click “Run Tool” to begin RAWR analysis. In the righthand bar, the submitted job will display green once the RAWR analysis has completed. A Newick tree with tree support values and/or MSA feature annotation text file will be saved as output, depending on the resampling task that was configured.
5. Other software tools in the Galaxy Tool Shed can be used to generate and display phylogenetic tree visualizations with support values. For example, the Newick Display tool can be downloaded and installed from the Galaxy Tool Shed (“Newick Display” under the repository name “newick utils”). Once installed, the Newick Display tool generates a phylogenetic tree visualization ( Figure 10).
6. After the visualization is generated, the “OpenSeadragon” tool can be used to display the image inside of Galaxy or saved to file. Figure 11 shows the relevant GUI workflow and Figure 12 provides an example of the resulting visualization within the Galaxy client.
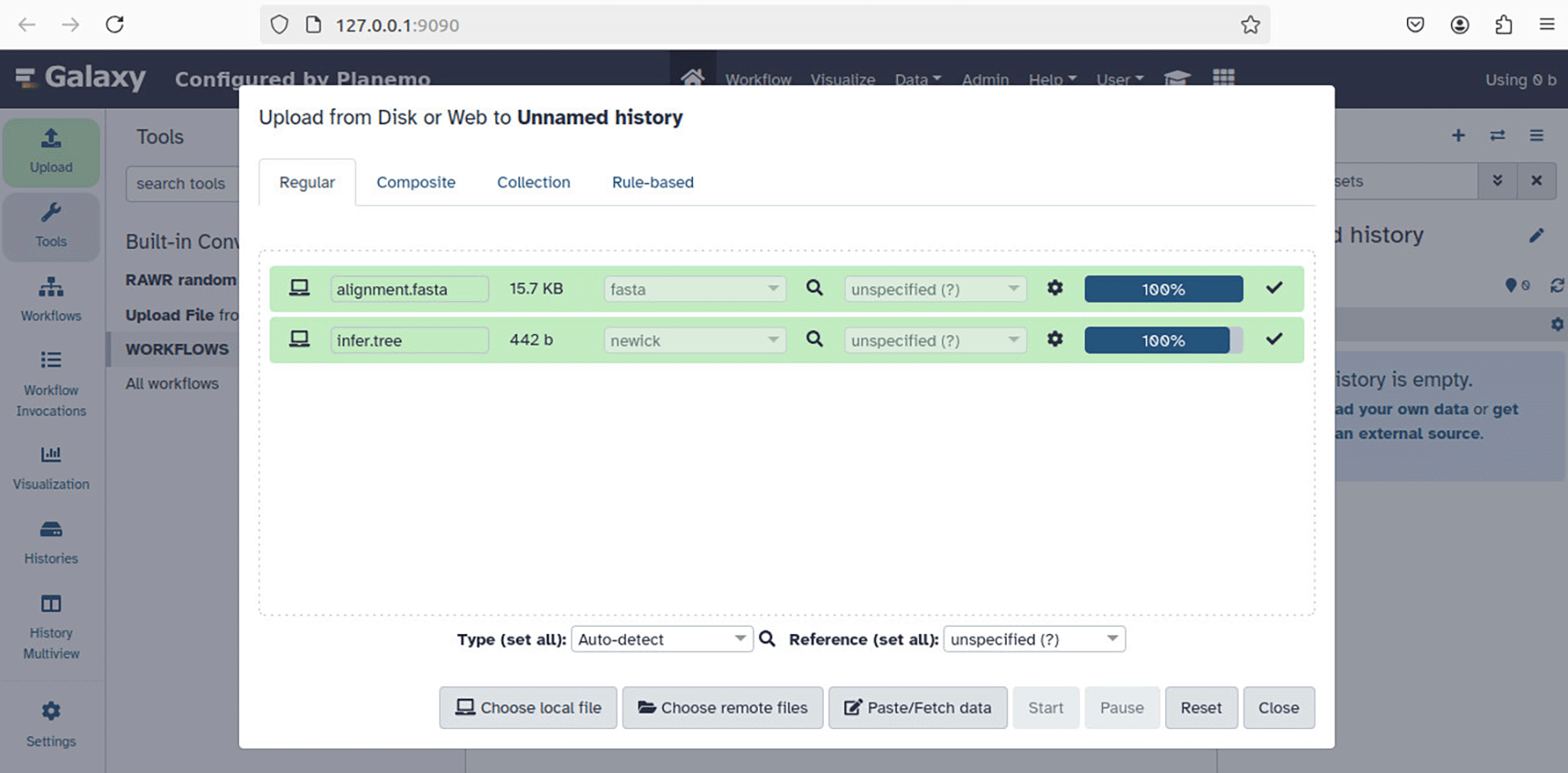
Input files for phylogenetic tree support estimation – “alignment.fasta” and “infer.tree” in the example as shown – correspond to an input MSA and annotation tree. After clicking “Start”, the input file upload begins and the client GUI dynamically updates based on the upload progress.
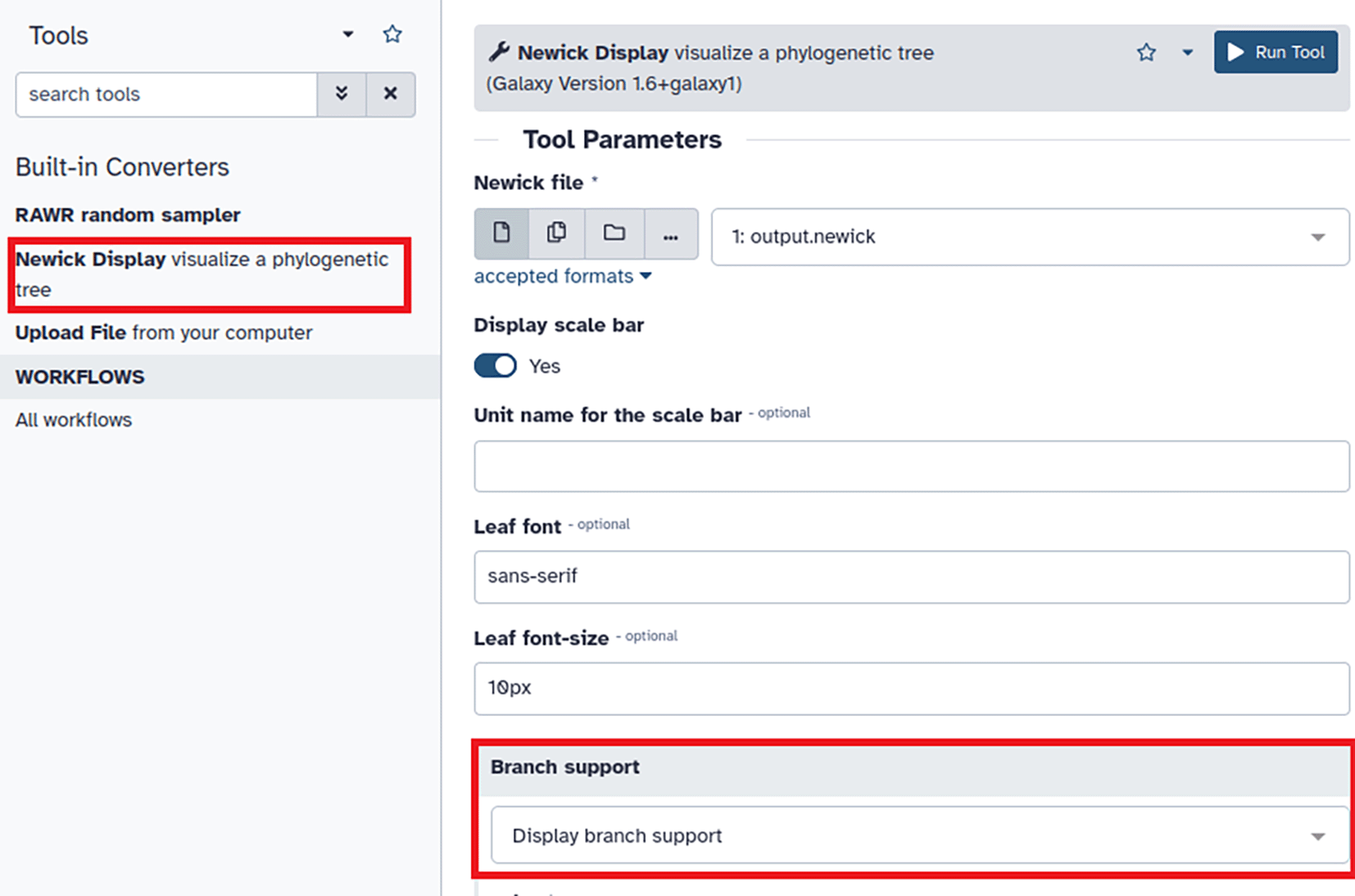
After RAWR analysis has completed, click “Newick Display” in the Tools pane (annotated with red rectangle in upper left of figure) and select “Display branch support” in the Branch support field of the Tool Parameters pane (annotated with red rectangle in lower left of figure). Then click the “Run Tool” button (upper right of figure) to generate image.
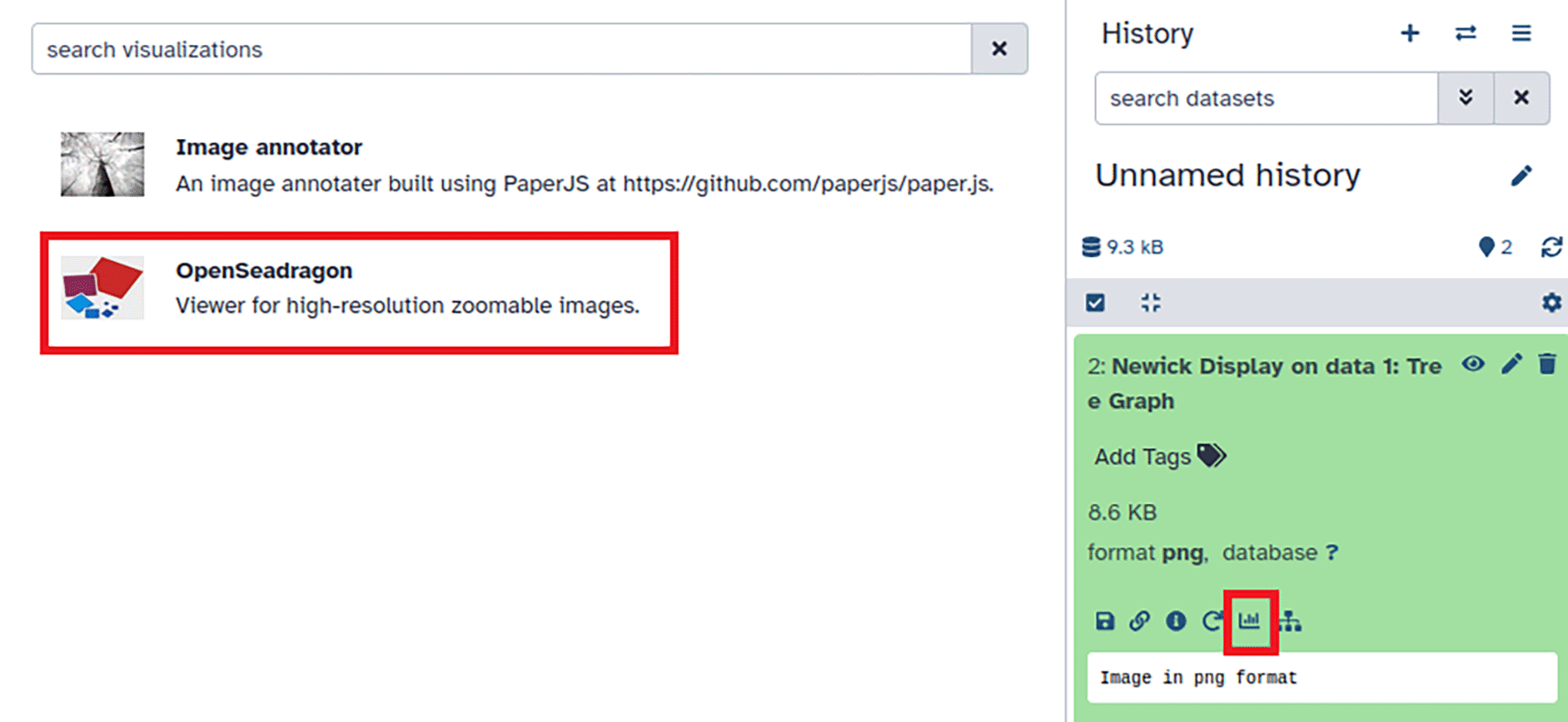
After generating a tree visualization using the Newick Display tool, click the visualization button (annotated with red square in lower right window pane) and select the “OpenSeadragon” tool (annotated with red rectangle in upper left of figure) to display the visualization.
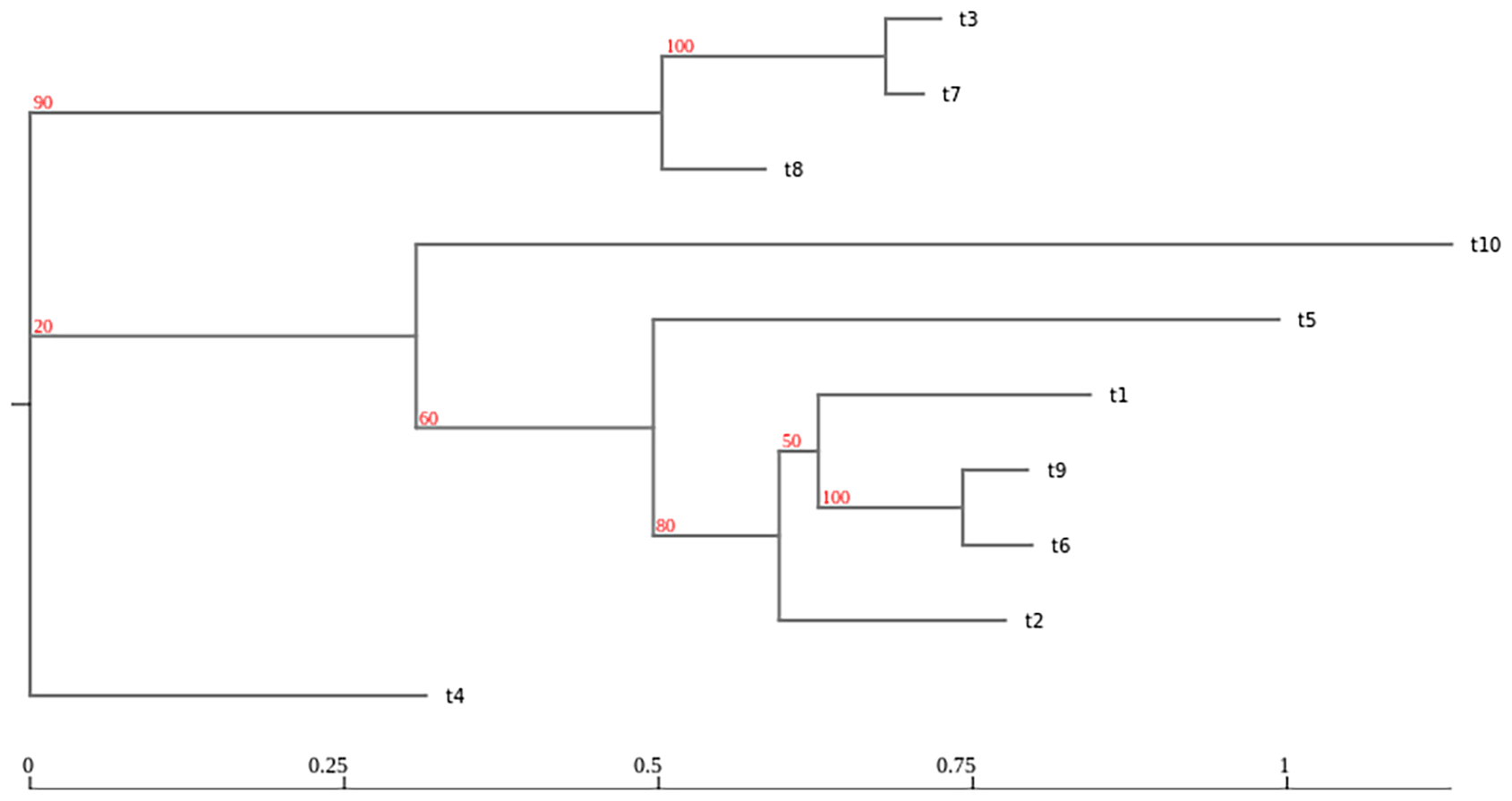
The results from a RAWR tree support analysis – i.e., the input annotation tree with RAWR support values annotating its branches – are visualized in the Galaxy client. RAWR support values are shown in red. The visualization was generated using the Newick Display tool and then displayed using the OpenSeadragon tool.
Web client. The following walkthrough uses the RAWR v1.0 web server to perform support estimation. RAWR analysis results are sent asynchronously via email.
1. Assuming that the web application has been set up in Anaconda, the conda environment is activated and the web server is started using the following commands.
$cd rawr-web $conda activate python3.10 $python app.py
2. The web server will be available locally at http://10.0.2.15:5000/. Any modern web browser can be used to access and interact with the web server. Figure 5 shows the main page of the web client.
3. Use the “Select Algorithm” drop-down menu to select an algorithm. By default, RAWR is chosen. Next, use the “Select Support Type” drop-down menu to configure the support estimation task – either phylogenetic tree support estimation or MSA support estimation. By default, phylogenetic tree support is selected. RAWR method parameters – i.e., the reversal probability γ and the number of resampled replicates – can be modified from default settings using the “Reverse Rate” and “Sample Number” fields, respectively.
4. Use a text editor to open an input file with a phylogenetic tree in Newick format, copy the tree’s Newick-formatted text string, and paste the text string into the “Phylogenetic Tree in Newick Format” text box.
5. Upload an MSA file using the “FASTA File Input” button.
6. Provide an email address in the “E-mail Address” field to receive results via email.
7. Click the “Submit” to execute the RAWR analysis.
8. Upon completion, the web page will be redirected to a download page. If phylogenetic support esti- mation is selected, a tree visualization will be provided ( Figure 13).
9. RAWR analysis results will also be sent asynchronously to the provided email address.
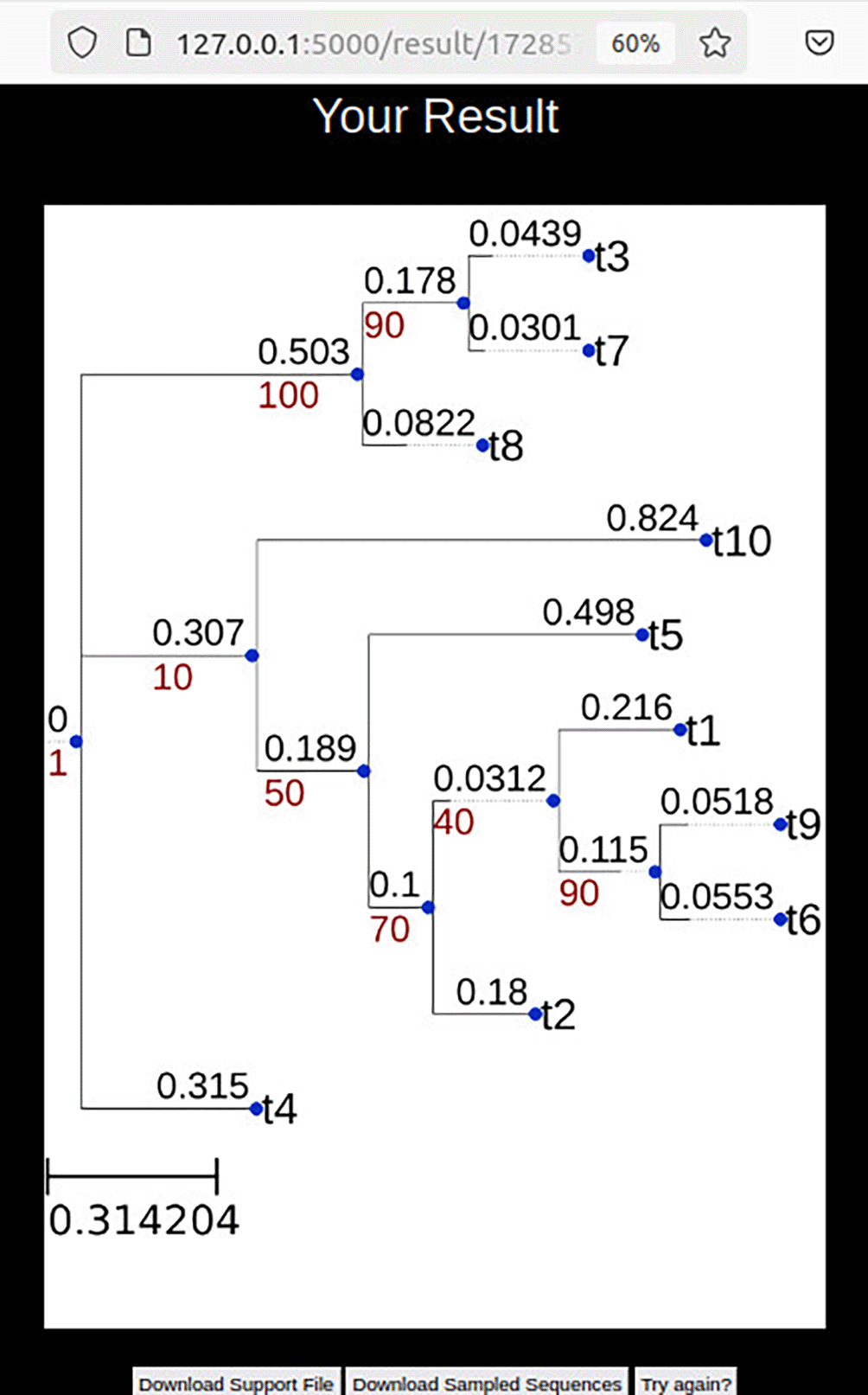
Once RAWR analysis concludes, the input annotation tree with RAWR support values is visualized. Output files can be downloaded. Results are also sent asynchronously via email if an email address was entered during analysis configuration.
Visualizing MSA support estimation results. RAWR-estimated MSA support values can be visualized in JalView (Waterhouse et al., 2009). The RAWR v1.0 software suite produces a feature annotation file that annotates the input MSA with RAWR support values. By default, the feature annotation file is named “MSA.support.csv jalview annotation.txt”. A JalView visualization can be produced using the input MSA file and feature annotation file as follows.
1. The input MSA file is specified by navigating to “File > Input Alignment > From File” ( Figure 14). An MSA visualizer window then appears.
2. The RAWR feature annotation file is input by navigating to “File > Load Features/Annotations” ( Figure 15).
3. A visualization of the input MSA with RAWR support values annotating MSA columns is displayed ( Figure 3).
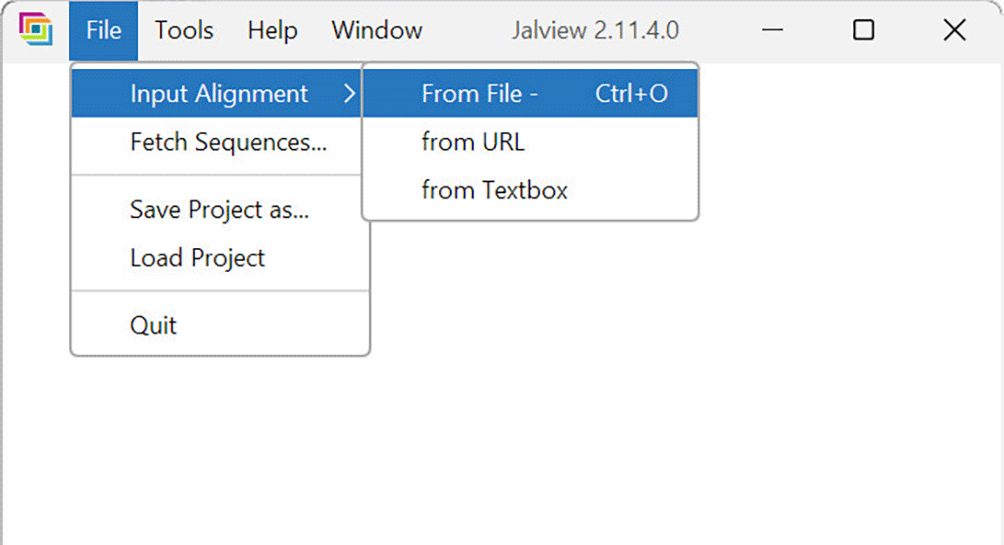
The desktop GUI for JalView v2.11.4.0 (Waterhouse et al., 2009) is shown.
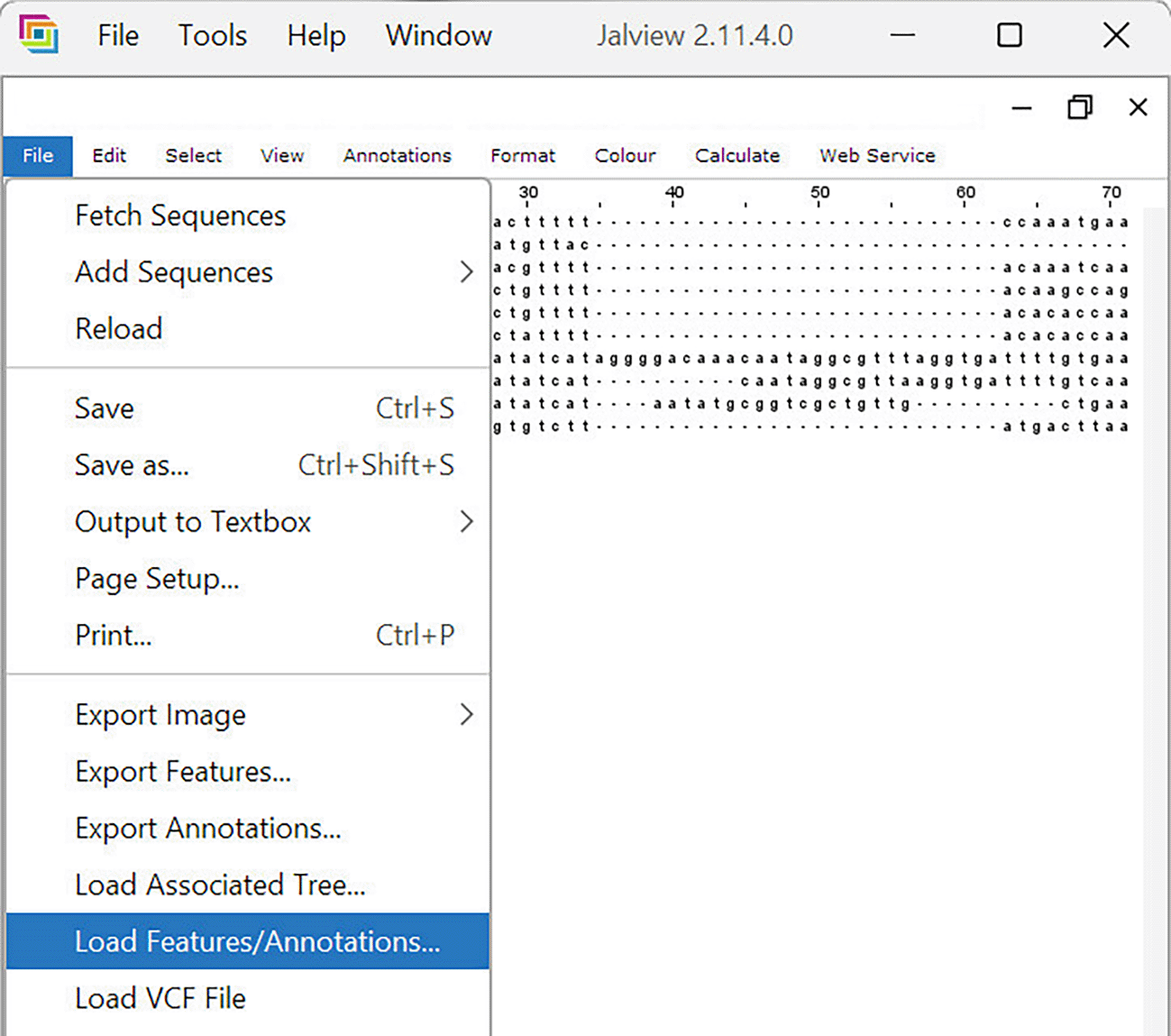
The desktop GUI for JalView v2.11.4.0 (Waterhouse et al., 2009) is shown. After an input MSA is loaded, the GUI updates with a dynamic MSA visualization. The RAWR-estimated feature annotation file is then loaded to annotate MSA columns with RAWR support values.
In summary, the RAWR v1.0 software distribution enables sequence-aware resampling of biomolecular and other sequence data, with applications to two primary resampling tasks – phylogenetic tree support estimation and MSA support estimation. User-friendly GUI clients are available for desktop PC, local Galaxy, and web software platforms. Developer support is also provided in the form of API access to software library functionality.
Future releases of the RAWR software distribution are anticipated in parallel with ongoing methodological research on sequence-aware resampling (Wang et al., 2020, 2021). The new software versions may include algorithmic enhancements to sequence-aware resampling techniques, new applications to classical or emerging resampling tasks, and new clients on existing and future computing platforms.
Source code available from: https://github.com/kjl-msu/RAWR-web-software.
Archived software available from: https://www.doi.org/10.5281/zenodo.18132473 (DOI: 10.5281/zenodo.18132473).
OSI approved open license software is provided under GNU General Public License version 3.
Supplementary Online Material document is available from Zenodo at https://doi.org/10.5281/zenodo.15391775 (DOI: 10.5281/zenodo.15391775). Materials have been posted under OSI approved open licenses (GNU General Public License version 3 and Creative Commons Attribution 4.0 International license).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)