Keywords
Chemokine signaling pathway, COVID-19, Cytokine storm, Gene co-expression network, Protein-protein interaction network
Chemokine signaling pathway, COVID-19, Cytokine storm, Gene co-expression network, Protein-protein interaction network
The SARS-CoV-2 infection has caused a global pandemic that impacted millions of people. Infection severity varies depending on individual characteristics, such as sex, lifestyle, and age.1,2 Disease severity is relatively higher in the senior population than in the younger population due to dysregulation in immune aging, which shifts immune cell responses toward inflammatory states and upregulates inflammatory gene expression. This dysregulation is linked to decreased antigen-presenting capacity, reduced heterogeneity of effector, cytotoxic, and fatigued CD28+ T cells, and age-associated B cells, impairing the adaptive immune system’s ability to limit infections and inflammation.3 Moreover, contrary to these findings, several young people suffer from severe lung diseases, indicated by hyperinflammation and low oxygen saturation,4,5 caused by dysregulation of cytokine release,6,7 known as a cytokine storm.
Cytokine storm is a condition that occurs when the immune system fails to eliminate a viral infection and subsequently promotes inflammation, along with an increase in the number of cytokines released.6,8 The endothelial cell lining of lung capillaries undergoes a state of inflammation caused by SARS-Cov-2 infection. This lung-triggered inflammatory response can spread from the site of infection into systemic circulation.9–11 This phenomenon demonstrates a possible interaction between the lungs and the bloodstream, in which pulmonary inflammation may influence and interact with the blood. Furthermore, the underlying cytokine storm mechanism is still under discussion as to whether it is the direct or synergistic effects of the virus.12,13 The investigation of the underlying cytokine storm mechanisms, especially the interplay between the lungs and bloodstream during a cytokine storm, enables the development of interventions to prevent the destructive effects of the cytokine storm that might reduce mortality rates. In addition, understanding lung-blood interactions can help prevent the long-term effects of cytokine storms and improve survivors’ overall quality of life by mitigating lasting health complications caused by severe inflammation.
Network-based analysis is a suitable approach for investigating disease mechanisms,14,15 especially for highly complex systems, such as the immune system. Nangraj et al. (2020) utilized network-based analysis, including a gene expression and protein-protein interaction network, to investigate the molecular mechanisms underlying esophageal adenocarcinoma.16 Gene Co-expression Network estimates the relationship between genes by calculating their correlation using gene expression value. The correlated genes are assumed to be functionally coordinated under certain conditions.17 Co-expression analysis has also been employed to study several infectious diseases, including influenza, tuberculosis, and hepatitis,18 illustrating the mechanistic interaction between the pathogen and host systems.
In this study, lung and blood transcriptomic gene co-expression networks were constructed to identify gene clusters (modules) that correlated with cytokine storms. Gene functional enrichment analysis was used to determine potential biological activities. The potential interplay between the lung and blood was explored using a protein-protein interaction network. Genes in the modules correlated with cytokine storms were used to construct the protein-protein interaction network. Furthermore, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway was employed to augment insights into the potential interaction between the blood and lung. This study provides valuable insights into the potential interaction between the lung and blood mechanisms of cytokine storms in COVID-19 patients. Understanding the interplay between the lungs and blood could provide a holistic view of the complexity of the molecular interactions involved in cytokine storms. Therefore, the insights gained from this research can positively impact the development of targeted therapies that address the systemic nature of cytokine storms, potentially leading to more effective treatment strategies for COVID-19 patients.
The analysis began with the retrieval of gene expression data from the Gene Expression Omnibus (GEO) database19 using specific keywords related to lung and blood tissues. Collected data without lung or blood gene count data, and the clinical conditions were excluded. The screened data underwent preprocessing before analysis. The preprocessed lung and blood data sets were used to construct separate gene co-expression networks for each tissue, followed by module detection. Essential lung and blood gene co-expression network modules were identified using module-traits correlation and module functional analysis. Module-trait correlation identified modules significantly associated with cytokine storm samples.20 Functional analysis of these modules explored the potential biological processes involved.
Genes from the identified essential modules were used to construct the protein-protein interaction (PPI) network. The biological significance of the network was analyzed using the Gene Ontology (GO) Biological Process and KEGG enrichment analysis. Gene Set Variation Analysis (GSVA) was employed to observe gene expression trends within the network.21–23 The potential interactions between the lung and blood tissues were explored based on the results. The entire analysis process is illustrated in Figure 1.
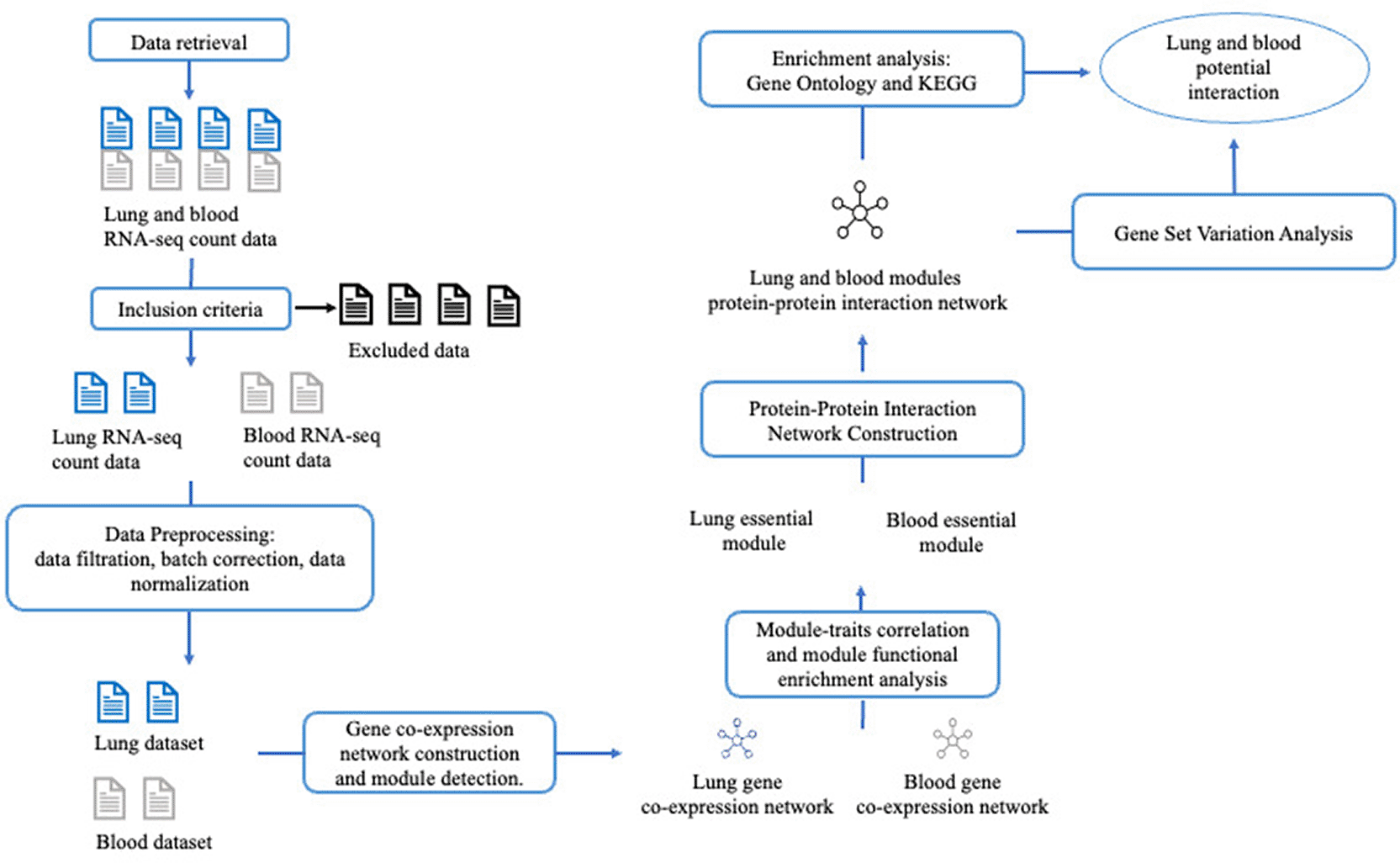
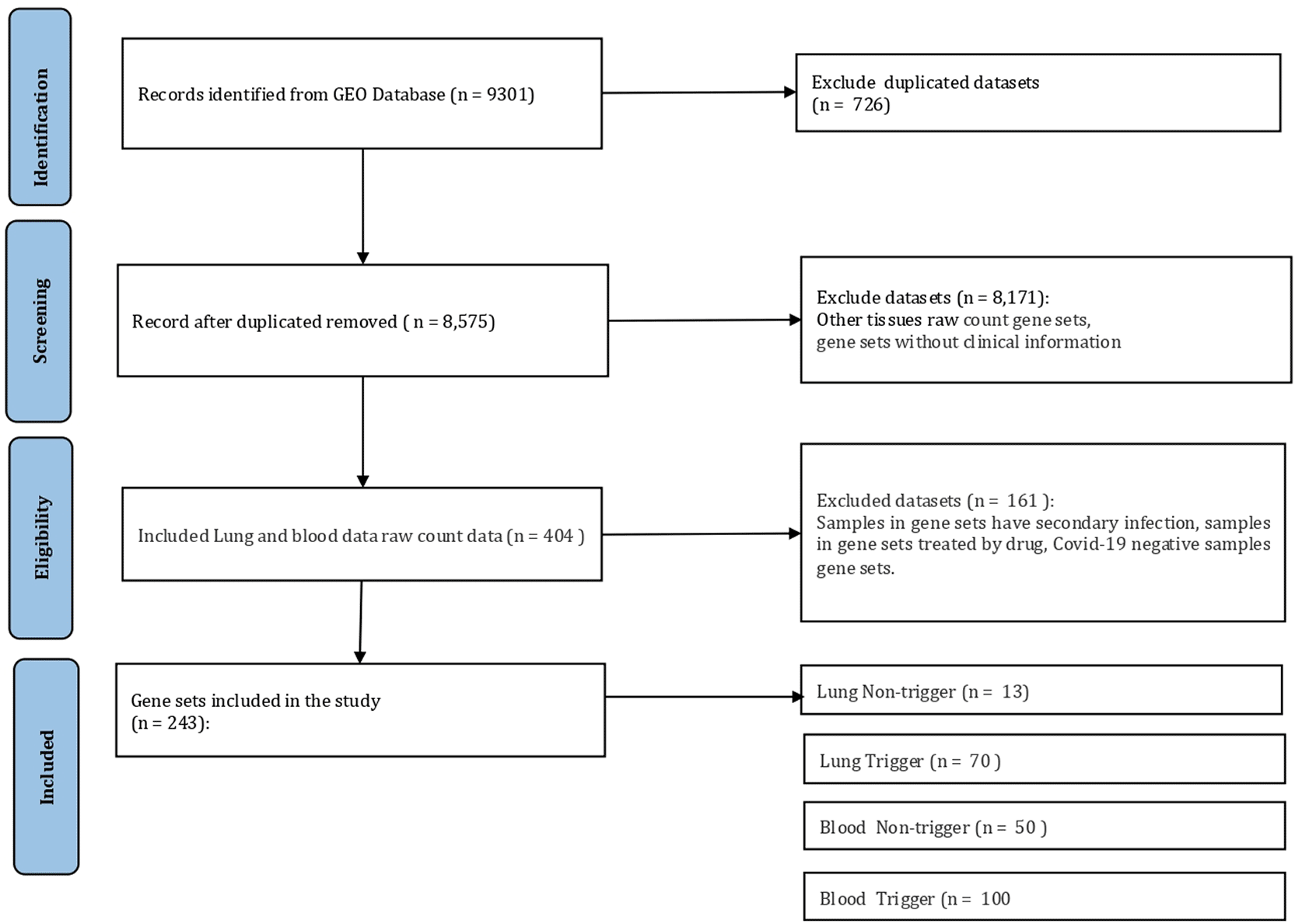
The ‘rentrez’ package24 was utilized to retrieve transcriptomic gene expression data from the GEO database.19 The series ids (GSE IDs) that specific to lung and blood tissues from patients diagnosed with COVID-19 were retrieved using ‘entrez_search’ command by keywords: “(lung tissue from COVID-19 patients) AND Homo sapiens [porgn:__txid9606] AND Expression profiling by high throughput sequencing [Filter]” and “(whole blood tissue from COVID-19 patients) AND H. sapiens [porgn:__txid9606] AND Expression profiling by high throughput sequencing [Filter]”. Duplicate GSE IDs and datasets that did not contain clinical information for the individual samples were excluded. The data collection process encompassed the retrieval of gene count data and sample clinical conditions from two sources: the GEO database and original publications. The data retrieval process is illustrated in Figure 2, and the sample level metadata is provided in Supplementary Table 1 (refer to extended data54). The data retrieval process was conducted on January 30, 2023.
Expression data was separated based on clinical data into two groups: ‘cytokine storm trigger,’ and ‘cytokine storm non-trigger.’ The non-trigger group consisted of positive COVID-19 samples obtained from the lung or whole blood tissue that did not exhibit any specific clinical conditions to cytokine storms, such as pneumonia, edema, dyspnea, hypoxemia, and acute respiratory distress syndrome (ARDS).25–27
Lung and blood datasets were processed independently using RStudio version 4.3.1,28 utilizing the sva (v3.52.0)29 and edgeR (v3.34.1)30 packages. First, gene counts were converted to counts per million (CPM) using the CPM function, and genes with expression levels below 10 CPM were excluded.31 Subsequently, batch effects were corrected using the ComBat_seq function from the sva package.32 Finally, the data were normalized using the Trimmed Mean of M-values (TMM) method described by Robinson et al.33
A gene co-expression network was used to identify the essential gene cluster (module) within samples associated with the triggering of cytokine storms. The construction of the lung and blood networks was carried out separately using gene expression data obtained from samples classified as ‘cytokine storm triggers’ and ‘cytokine storm non-triggers’.
Network construction was accomplished using the Weighted Gene Co-expression Network Analysis (WGCNA)20 package version 1.72–5 in R. The ‘pickSoftThreshold’ function was employed to determine the soft threshold for ensuring the scale-free topology network prior to network construction. Subsequently, the connection between each pair of nodes is calculated using an adjacency matrix with a determined soft threshold as the weighting factor. The adjacency matrix is subsequently employed in the computation of the Topological Overlap Matrix (TOM). The gene cluster (module) was detected using hierarchical clustering with TOM-based dissimilarity. The ‘cutreeDynamic’ function, with a minimum module size of 30, partitioned genes with similar expression profiles into distinct modules, each assigned a unique color for identification.16,18,20
Module eigengenes, which represent the principal component of each module, for each module were calculated using WGCNA’s ‘moduleEigengenes’ function. The calculation and identification of potential essential modules were based on the association between module eigengenes and cytokine storm samples16,20 to identify significant associations. The ‘bicor’ function of the WGCNA package was utilized to calculate the correlations, helping to identify potential essential modules linked to cytokine storm triggers.20,34
The gene enrichment analysis was performed in R using the ‘anRichment’ package version 1.26 to gain insight into the biological functionality of each module.20,35 The process of enrichment analysis was performed using the function ‘enrichmentAnalysis’. The function requires two primary inputs: classes (network modules) and a compilation of reference gene sets (GO biological process gene sets). The reference collection must be generated before analysis by retrieving all the gene sets from GO using the ‘buildGOcollection’ function and then selecting the biological process gene sets with the ‘subsetCollection’ function.20,35 This analysis provided insights into the biological functionalities of the identified modules.
Essential modules were identified based on the following criteria: a correlation value greater than 0.2, a p-value lower than 0.05, and the presence of Gene Ontology (GO) terms associated with the immune system process.20,36 The potential interaction between lung and blood important modules was investigated using a PPI network. Construction of the PPI network was facilitated using the STRING database.37 The gene member associated with significantly enriched terms from each module was utilized as an input for constructing the protein-protein interaction network. The network was constructed utilizing multiple sources of interaction, including experimental data, databases, and co-expression. The confidence interaction score used was 0.9 to reduce the number of false-positive interactions.16,38 The constructed network and the Gene Ontology (GO) cell component associated with each node were exported for further processing using Cytoscape version 3.10.1.39
The constructed PPI was processed using Cytoscape. The GO cell component of the node and the information regarding the membership of genes in lung or blood modules were imported into Cytoscape. The GO cellular component annotation is utilized to determine the subcellular localization of the protein encoded by the corresponding genes. The hypothetical interaction between lung and blood genes, which is biologically implausible, was eliminated from consideration. The exclusion criteria refer to the interactions between lung genes responsible for producing cytosolic proteins and blood genes responsible for producing extracellular or membrane proteins, and vice versa. Following this, the largest subnetwork containing interactions between lung and blood genes was chosen.
Using the string enrichment tool within Cytoscape facilitated the identification of the biological processes linked to the network.39,40 ShinyGO version 0.80 was employed to obtain further understanding regarding potential KEGG pathways that establish a connection between the interaction of the lung and blood.21
The Gene Set Variation Analysis (GSVA) package in the R programming language was utilized to determine the gene expression pattern derived from the gene members within the network. A list of network members was used as the gene set. The ‘gsva’ function was used to calculate the enrichment score of a gene list for each sample.22 The calculated enrichment scores of cytokine storm samples and cytokine storm non-trigger samples were then compared to observe the gene set activity.23
The specified keywords were used to retrieve gene expression data from the GEO database, resulting in 9,301 gene expression data, including 726 duplicated data. Gene expression data unrelated to the lung or blood and data lacking clinical information about the sample were excluded. This process resulted in a final set of 404 gene expression data. Furthermore, 161 data were excluded since the sample had a secondary infection or had been treated with a drug. Finally, 243 gene expression data were included in the study and classified into cytokine storm trigger and cytokine storm non-trigger. There are 13 lung samples classified as non-trigger for cytokine storms, 70 lung samples as cytokine storm trigger group, 50 blood samples as non-trigger for cytokine storms, and 100 blood samples as cytokine storm triggers from blood. The lung data set consists of 16,776 genes per sample, while the blood data set consists of 19,047 genes.
The construction of the gene co-expression network involved the utilization of predetermined soft thresholds, specifically 8 for lung samples and 3 for blood samples. The soft threshold was chosen to maintain a scale-free network structure while preserving a high level of connectivity.20 The Topological Overlap Matrix (TOM) dissimilarity-based hierarchy clustering method was employed to identify modules within the network, and there are 28 modules in the lung and 29 modules in the blood datasets. The identified modules were then assigned into colors.16,18,20
Twelve blood modules, including light cyan, dark magenta, pale turquoise, tan, turquoise, orange, royal blue, sky blue, sky blue 3, dark grey, cyan, and purple, have a significant positive correlation with cytokine storm trigger samples. Furthermore, seven modules, blue, dark turquoise, light pink 4, violet, dark magenta, grey, and light grey, were found to have a significant positive correlation with cytokine storm trigger samples in the lung. The module correlation value is illustrated in Figure 3.
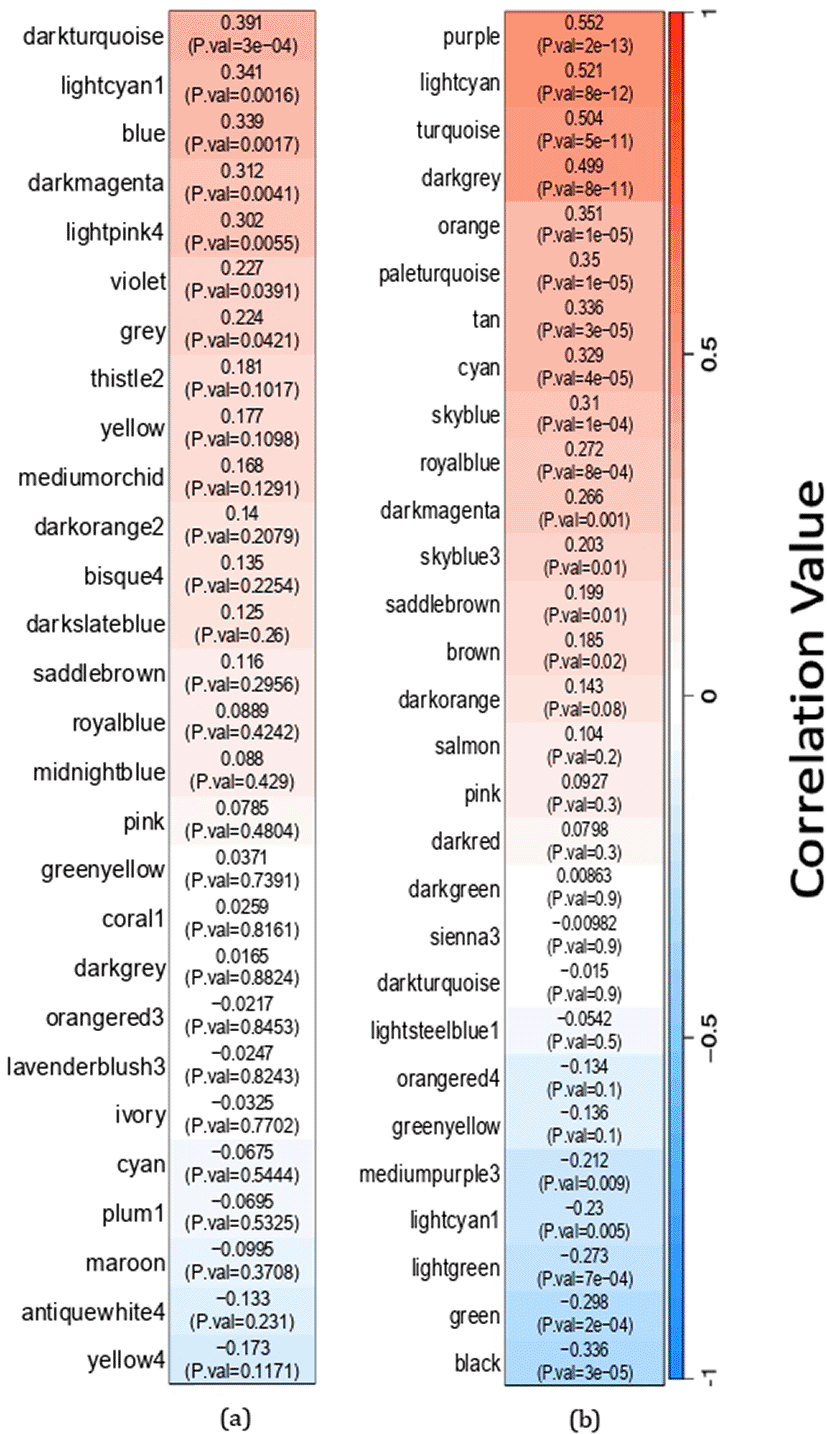
The heat map demonstrates the correlation between modules and cytokine storm trigger samples. Every cell presents the respective correlation coefficient and p-value. The red squares represent a module with a positive correlation with storm trigger samples, while the blue squares indicate a module with a negative correlation with storm trigger samples.
The module was evaluated by enrichment analysis utilizing the Gene Ontology (GO) Biological Process dataset to enhance understanding of its biological function. The enrichment result was used to consider which modules are essential to cytokine storm samples. Figure 4 visualizes the number of significantly enriched GO Biological Process terms that are positively correlated with cytokine storm triggers in both blood and lung modules. The analysis indicates the blood has four modules with immune system-related GO terms, including tan, sky blue, sky blue 3, and dark grey, whereas the lung only has one, which is dark turquoise. The top 10 significant enriched terms for each module are visualized in Supplementary Figure 1. According to the GO Biological Process, the module involved in the immune system process will proceed for further analysis to identify the cross-tissue module interaction.
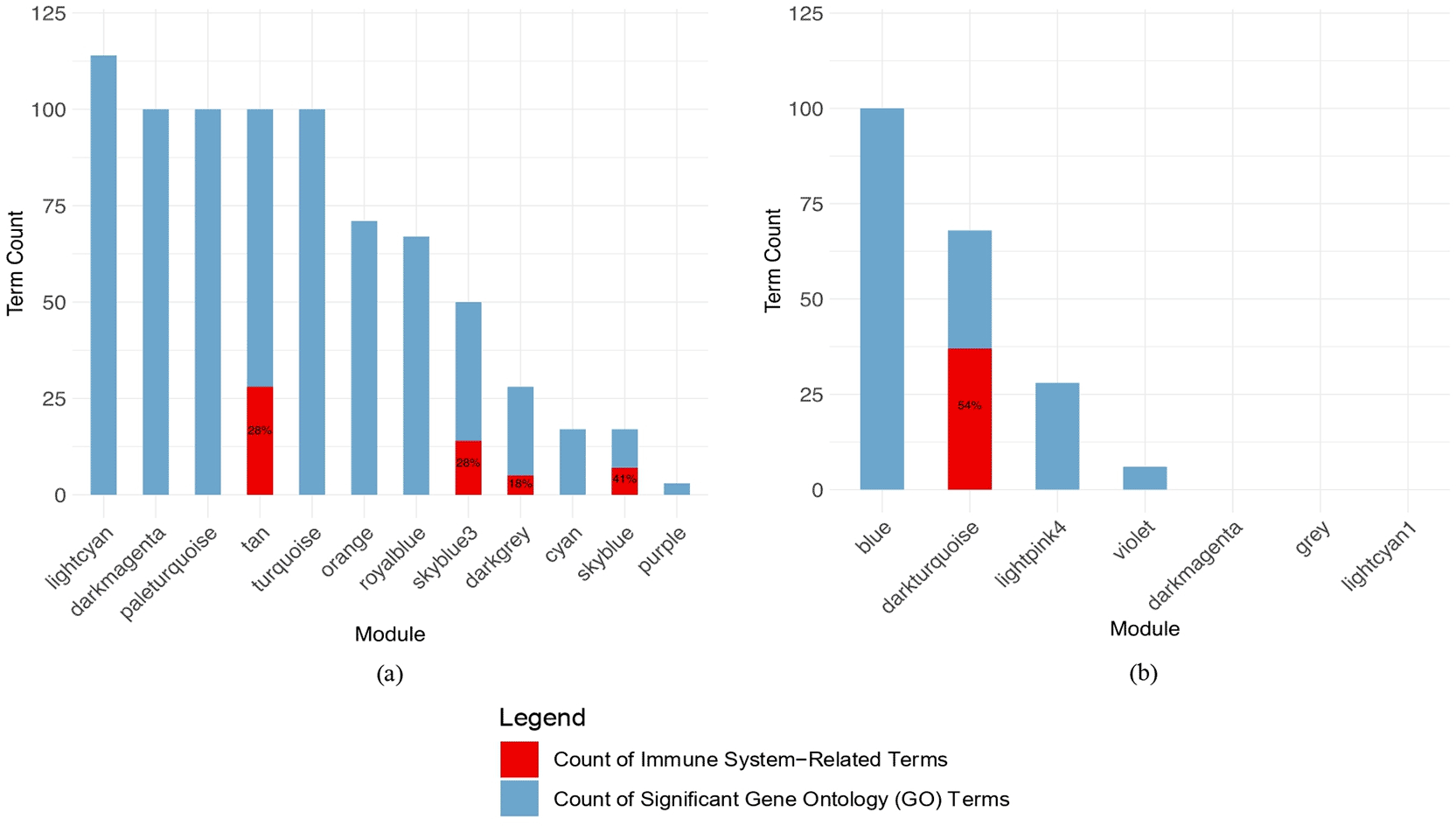
The count of significantly enriched Gene Ontology terms within the parent terms “Biological Process” category for each module is depicted using a blue bar. The red bar indicates the count of Gene Ontology terms that are considerably enriched within the parent term “Biological Process” category, specifically under the child term “immune system process”. The ratio of immune system process-related terms to the total number of biological process terms is shown in percentage within the graph.
The gene members of the top 10 enriched terms from each selected module were utilized to investigate the potential interaction between lung and blood modules. Four hundred sixty-seven genes were used as input to the String database for constructing a protein-protein interaction network. The physically infeasible link between cytosolic lung protein and blood cytosolic protein was excluded, and the largest subnetwork is depicted in Figure 5. At the same time, the whole network is visualized in Supplementary Figure 2.
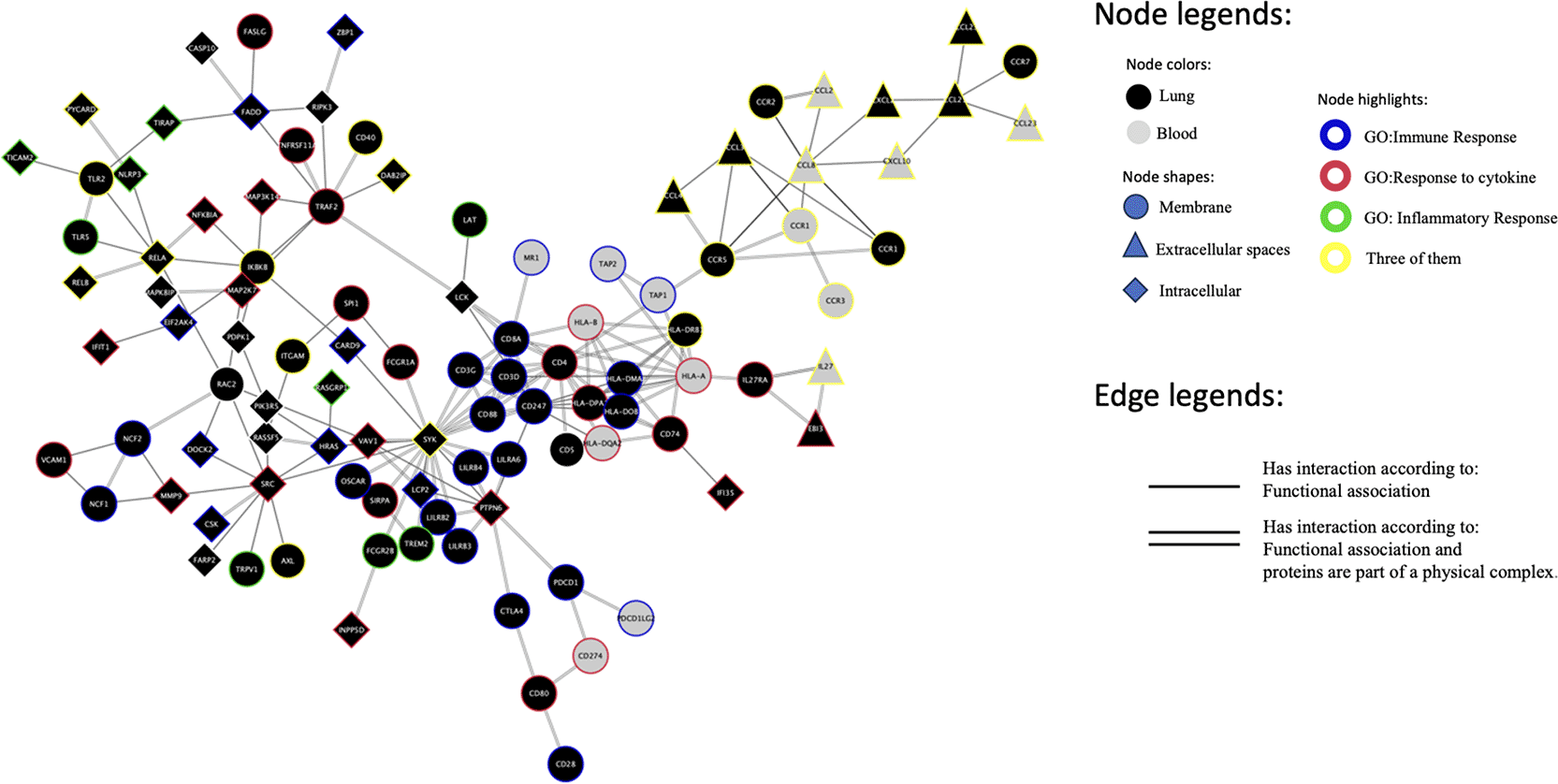
The black nodes represent lung module genes, while the grey nodes illustrate blood module genes. The node shapes indicate the gene’s protein product location. Circle nodes represent cell membrane protein, triangles external protein, and diamonds intracellular protein. Gene membership to GO biological process terms is also highlighted with several colors for nodes. Gene Ontology (GO) immune response term nodes are highlighted in blue. Red highlighted nodes represent GO response to cytokine term members. Green highlighted nodes are GO inflammatory response members. Lastly, the nodes marked with yellow represent genes associated with membership in all three categories, as mentioned above. One-line edges imply functional association, while the two-lines show that the proteins interact according to functional association and can create physical complexes.
The protein-protein interaction network was then enriched with the gene’s GO biological process membership. The results demonstrate that genes from the lung and blood potentially interact and are involved in immune responses related to inflammation and cytokine response. Following this, the KEGG pathway information was utilized to identify the possible pathway interaction within the subnetwork. The analysis demonstrates potential lung and blood module interaction in the KEGG chemokine signaling pathway, followed by NfkB signaling pathway activation, as visualized in Figure 6.
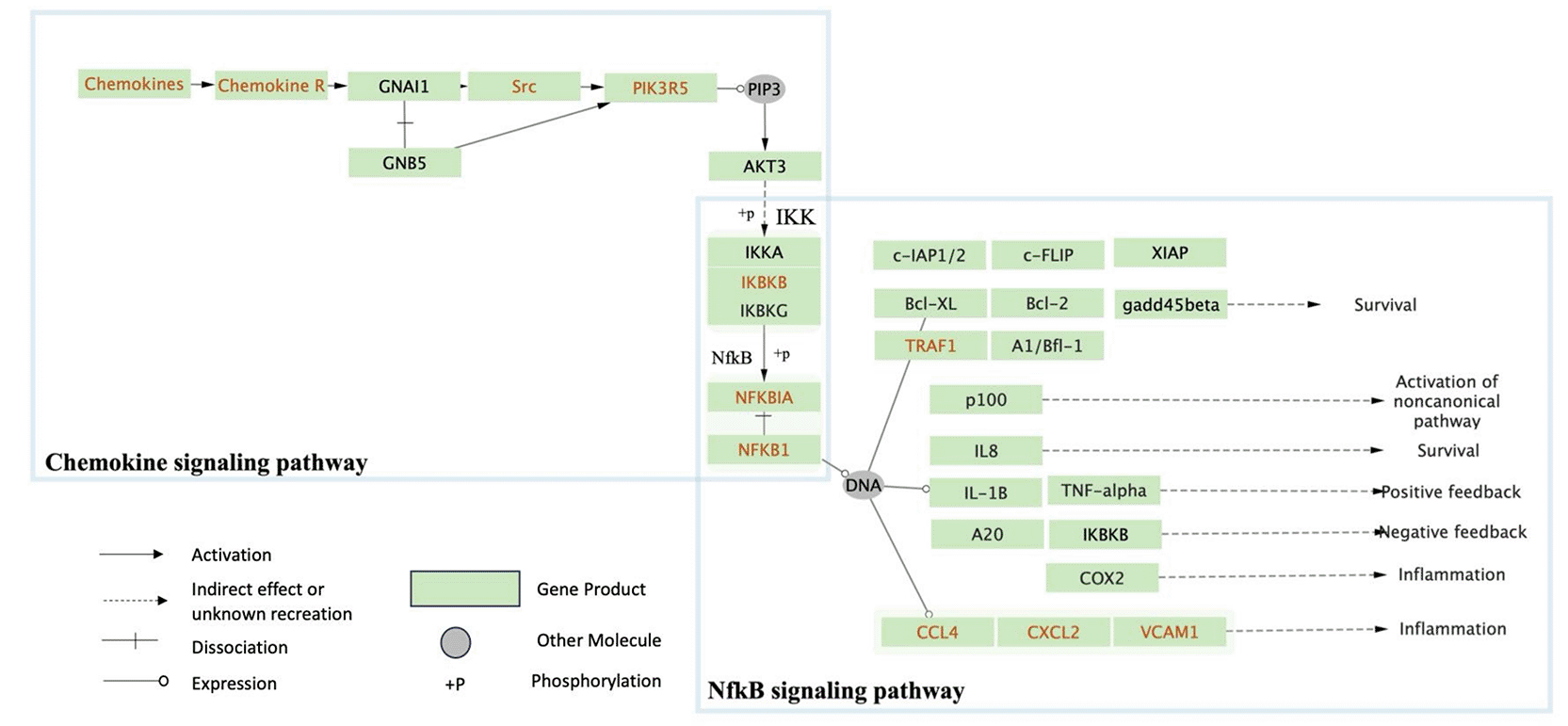
The genes that have been mapped within the pathway are indicated in red.
The gene expression trend of the network was evaluated by calculating the GSVA enrichment score. GSVA enrichment score assesses the variation of gene set expression pattern for each sample regardless of the sample label. The enrichment score demonstrates the degree of gene set coordinated activity. The GSVA enrichment score was implemented in this work to assess the gene expression pattern in lung and blood samples relative to the overall expression observed in each dataset.22 Figure 7 demonstrates that the GSVA enrichment score for the lung cytokine storm trigger samples increased significantly from −0.28 to 0.01 compared to the lung cytokine storm non-trigger samples under the p-value less than 0.05. Similarly, the enrichment score of blood cytokine storm trigger samples increased significantly from −0.26 to 0.02 compared to the blood cytokine storm non-trigger samples under the p-value less than 0.05. The significantly increased enrichment score suggests that the gene set activity is increased,22,23,41 indicating that the network activity is upregulated in cytokine storm trigger samples compared to the cytokine storm non-trigger samples.
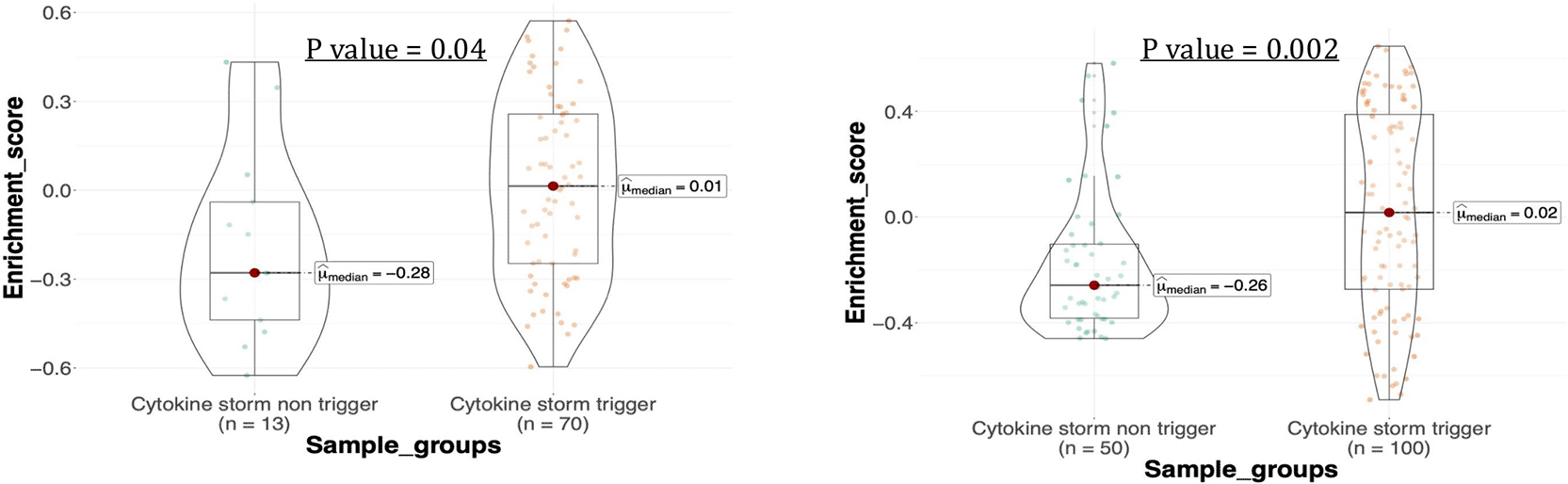
(a) Lung, (b) Blood.
The count of significantly enriched Gene Ontology terms within the parent terms “Biological Process” category for each module is depicted using a blue bar. The red bar indicates the count of Gene Ontology terms that are considerably enriched within the parent term “Biological Process” category, specifically under the child term “immune system process”. The ratio of immune system process-related terms to the total number of biological process terms is shown in percentage within the graph.
The black nodes represent lung module genes, while the grey nodes illustrate blood module genes. The node shapes indicate the gene’s protein product location. Circle nodes represent cell membrane protein, triangles external protein, and diamonds intracellular protein. Gene membership to GO biological process terms is also highlighted with several colors for nodes. Gene Ontology (GO) immune response term nodes are highlighted in blue. Red highlighted nodes represent GO response to cytokine term members. Green highlighted nodes are GO inflammatory response members. Lastly, the nodes marked with yellow represent genes associated with membership in all three categories, as mentioned above. One-line edges imply functional association, while the two-lines show that the proteins interact according to functional association and can create physical complexes.
The phenomenon known as cytokine storm has drawn significant attention concerning the novel Coronavirus disease 2019. Patients infected by this disease exhibit elevated levels of several pro-inflammatory cytokines, which are associated with the severity of the illness.42 The term “cytokine storm” was initially used in 1993 to characterize the occurrence of acute graft-versus-host diseases in the context of engraftment syndrome after allogeneic stem-cell transplantation.43 It refers to a group of hyperinflammatory immune dysregulation disorders characterized by systemic inflammation, and multiorgan dysfunction, leading to potential multiorgan failure and death. These hyperinflammatory disorders include pathogen-induced, neoplasia-induced, monogenic, and iatrogenic causes,44,45 all featuring extensive cytokine release resulting from the overstimulation of immune cells. This dysregulated cytokine release also significantly impacts the hematopoietic system and hemostasis,45,46 suggesting an interaction between the infected site, the lung, and the circulatory system.
This study investigates the potential interaction between lung and blood tissue in COVID-19 patients experiencing cytokine storms. The gene co-expression approach was utilized to evaluate the expression patterns in patients who triggered cytokine storms and those who did not. Constructing gene co-expression networks provides valuable insights into how specific genes and their interactions contribute to the cytokine storm response in COVID-19 patients. Genes with similar expression patterns were clustered into modules, suggesting coordinated responses to the disease and potential interactions among these genes.47 This foundational step helps unravel the molecular mechanisms underlying cytokine storms in COVID-19.
Identifying important modules related to cytokine storm samples was achieved by calculating the correlation values between modules and cytokine storm trigger samples. Modules with positive correlation values potentially play roles in disease progression.48 Building on the established definition of a cytokine storm as immune system dysregulation, modules enriched with GO terms related to the immune system were prioritized for investigation. This prioritization strategy resulted in the identification of four blood modules and one lung module suggesting that these modules were likely involved in cytokine storm in COVID-19 patients. A PPI network was employed to explore the potential interaction between the selected lung and blood modules.
The PPI network models the gene interaction within the selected modules via its protein product, being a key in cellular communication.49 By examining the closely interacting genes within the network, it is possible to gain more insight into cellular communication. The constructed network enriched by the GO biological process information demonstrates the interaction between blood and lung module genes. GO biological process terms such as “inflammatory response,” “response to cytokine,” and “immune response” were identified within this enriched subnetwork. These terms signify the biological processes that these genes are collectively involved in. The findings indicating a potential interaction between cytokine genes from the blood module and cytokine receptors genes from the lung module, and vice versa, provide a tangible example of how these modules actively communicate with one another and may have essential roles in processes related to inflammatory and immune responses.
Pathway information from KEGG was utilized to find the specific lung and blood module interaction and its potential effect. The examination of KEGG pathway information revealed a potential link between the lung and blood modules, primarily orchestrated by KEGG chemokine signaling. Notably, interactions between specific genes, such as lung CCL3 and blood CCR1, blood CCL2 and lung CCR2, and lung CCL8 and blood CCR5, CCR1, and CCR2, highlight dynamic cellular communication. These chemokine-receptor interactions collectively activate NfKB via src activation, triggering PI3k and Akt phosphorylation to IKK, and initiating an inflammatory response.
The network members’ gene expression trend was examined by calculating the GSVA enrichment score, representing the integrated level of gene set expression higher the GSVA score indicates greater overall expression of the gene set in the studied group.27 Interestingly, there is an increasing trend in gene expression profiles within the network when comparing trigger and non-trigger group samples. This increase suggests heightened activities between lung and blood module genes, which may contribute to overwhelming inflammation observed in patients with cytokine storms, as the genes within the network have a role in the inflammatory response. The findings align with the studies on post-mortem lung samples of COVID-19 patients showed a significant increase in chemokine production, including CCL2, CCL8, and CCL11 genes, which was connected to a strong and ongoing inflammation during the early stages of infection and prolonged intensive care unit (ICU) stays.50
The results suggest that the increasing chemokine signaling interactions between lung and blood may be critical in deteriorating the patient’s condition. SARS-CoV-2 infection leads to lung endothelial cell activation, which releases chemokines from the lung to the circulation system.51,52 The released chemokines then bind their receptors on circulating monocytes and macrophages, recruiting them to the site of infection. The recruited macrophages initially express high levels of chemokines inducing vascular leakiness.50,53 Additionally, the activated macrophages induce excessive fibroblast activation, leading to the onset of pulmonary fibrosis and respiratory failure.53 Therefore, the chemokine signaling between the lung and blood is a potentially vital interplay in the deterioration of the patient’s condition. Nevertheless, it is important to acknowledge certain limitations. This study grouped samples into cytokine storm trigger and non-trigger based on available clinical information, leading to the exclusion of some datasets. Addressing this limitation is essential for enhancing the robustness of the results. Despite this, the findings support prior studies, emphasizing the critical role of chemokine signaling in cytokine storms and suggesting a possible interplay between the lung and blood during the disease progression.
The research investigates the potential interaction between lung and blood tissue that triggers cytokine storm in COVID-19 patients. By utilizing gene co-expression and protein-protein interaction networks, the study identified gene clusters within specific modules in blood and lung samples that show dynamic interaction during cytokine storms. The observed interaction is linked to the excessive inflammatory response observed in individuals experiencing cytokine storms, as evidenced by the increasing expression of genes within the network. The findings suggest that the escalating chemokine signaling activity between the pulmonary and circulatory systems plays a pivotal role in the deterioration of the patient’s condition. This work emphasizes the importance of chemokine signaling in cytokine storms and highlights the potential interaction between the lung and blood during the disease progression, while also acknowledging limitations such as the availability of clinical data. Further research and empirical evidence are necessary to confirm these findings and to deepen our understanding of COVID-19-related cytokine storms.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)