Keywords
Virus of herpes zoster, Varicella-zoster virus, pathogenicity, genes for virulence, immunity.
This article is included in the Fallujah Multidisciplinary Science and Innovation gateway.
This article is included in the Pathogens gateway.
Virus of herpes zoster, Varicella-zoster virus, pathogenicity, genes for virulence, immunity.
Shingles or herpes-zoster (HZ), results when the silent varicella-zoster virus (VZV), once re-activated in the sensory ganglia, again becomes active. HZ most typically appears as an acute uni- or bi-dermal dermatomal vesicular eruption with the addition of neurotic pain, with or without chronic post-herpetic neuralgia (PHN).1 VZV’s neurotropism and its advanced arsenal for evading immunity during lytic growth as well as during latency are repeated during reactivation. These include mechanisms for amplifying persistence in the neuron with explosive recrudescence by the peripheral nerves by kinases on disruption of innate signalization (ORA47/ORF66), direct inhibition by VZV on perception by the cytosol of DNA by inhibition by ORF9 on the cGAS pathway, with depression of antigen presentation (e.g., inhibition by VZV of the MHC pathway).1 Clinically, a painful, band-like vesicular rash that respects the midline is typically diagnosed at the patient’s bedside; however, laboratory confirmation is necessary for unusual, pre-eruptive, oral, ocular, or immunocompromised presentations. Polymerase chain reaction (PCR) is the preferred diagnostic method for VZV DNA from lesion material, preferably vesicle fluid, scabs, or cells scraped from the lesion base, and, when necessary, from sterile sites like cerebrospinal fluid in suspected neurologic disease; viral culture is slow and insensitive, and serology is not very useful for acute diagnosis. In addition to verifying infection, rapid, precise diagnosis directs early antiviral treatment to lower acute pain and PHN risk and stimulates assessment for neurologic, ophthalmic, or widespread consequences in high-risk hosts.2–4 Recent pathogenic insights from human neuronal models further clarify how VZV re-uses its immune-evasive behaviors in the nervous system, including reshaping autophagy, altering stress-granule dynamics, and reducing interferon programs. This links the clinical spectrum of HZ and its complications to molecular virulence strategies. Recent pathogenic insights from human neuronal models further clarify how VZV re-uses its immune-evasive behaviors in the nervous system, including reshaping autophagy, altering stress-granule dynamics, and reducing interferon programs. This links the clinical spectrum of HZ and its complications to molecular virulence strategies.1 The main topics covered in this review are the herpes zoster virus’s epidemiology, pathophysiology, clinical presentation, diagnosis, treatment, and prevention.
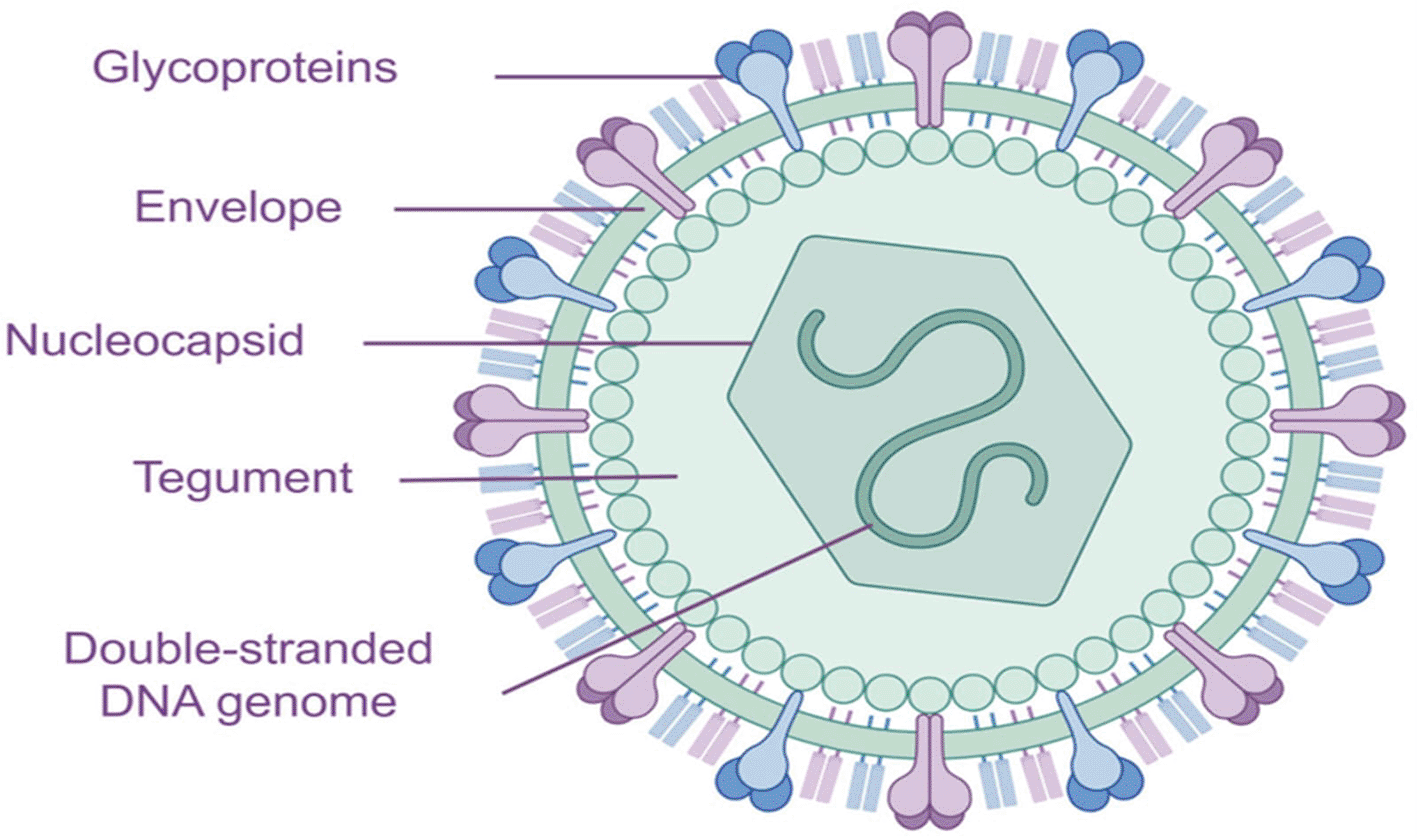
Varicella-zoster virus (VZV), also referred to as Human Herpesvirus 3, or HHV-3, is a highly common and clinically significant pathogen that is a member of the genus Varicella virus, subfamily Alphaherpesvirinae, and family Herpesviridae. It shares the closest kinship with the human herpes simplex viruses HSV-1 and HSV-2. Like all herpesviruses, VZV virions are complex, enveloped particles made up of four concentric structural layers: an icosahedral nucleocapsid encasing a central, linear, double-stranded DNA genome (~125 kilobases); a proteinaceous tegument layer made up of viral proteins and mRNAs; and a lipid bilayer studded with viral glycoproteins that are derived from host-cell membranes as shown in (Figure 1).5 With 162 capsomers (150 hexons and 12 pentons) and 20 triangular faces, the capsid takes on the distinctive herpesvirus architecture, which makes geometric assembly and DNA packaging easier. The tegument that surrounds the capsid has several functions, including the provision of elements required for early infection, the regulation of host reactions, and the facilitation of capsid transit into the cell.6,7 Multiple glycoproteins, including gB, gC, gE, gH, gI, gK, and gL, are densely decorated on the outer envelope and mediate host-cell receptor binding and membrane fusion during viral entry. Characteristically, VZV lacks the glycoprotein D (gD) present in HSV but instead depends on gB and the gH-gL heterodimer as its major fusogenic machinery. Studies on the VZV capsid (A-capsid) using cryo-electron microscopy (cryo-EM) at ~3.7 Å resolution have shown that, while its general design is similar to other herpesviruses, but it also possesses structural subtleties like assembly pockets and inter-capsomere interactions that could help antiviral targeting.8
Etiologic agent of herpes zoster (HZ, shingles), varicella-zoster virus (VZV), causes an appreciable and age-disproportional world disease burden. In many populations, the lifetime risk is approximately 20–30%, and the incidence increases sharply after age 50 as cell-mediated immunity declines, meaning that older adults and immunocompromised individuals account for the majority of cases and complications like post-herpetic neuralgia (PHN). HZ incidence in the general adult population is estimated by current, population-based estimates to be between 3 and 5 cases per 1,000 person-years in high-income settings, with regional and age-specific heterogeneity (older cohorts typically experience substantially higher rates).10 The local burden of immunosuppressive conditions and treatments, variations in health care access and diagnostic ascertainment, the prevalence of VZV seropositivity (which determines the pool at risk for reactivation), demographic structure, and the indirect effects of varicella (chickenpox) vaccination policies that can change opportunities for exogenous immune boosting of adults are the main causes of epidemiologic heterogeneity.11 The adjuvanted recombinant zoster vaccine (RZV, gE-based) shown very high efficacy in pivotal trials (≈90–97% against HZ) and consistently strong real-world effectiveness (usually reported in the 70–90% range depending on study design, age, and follow-up), significantly reducing incidence and severe outcomes where uptake is sufficient.12 These developments represent significant advancements in primary prevention of HZ over the past ten years. Although many national immunization schedules have only recently (or not yet) included routine HZ vaccination for older adults, uptake is still uneven due to programmatic, financial, and supply constraints; as a result, cost-effectiveness analyses and implementation studies continue to be crucial in determining policy.13 Standardized case definitions and, more recently, whole-genome sequencing of circulating VZV strains have been used in epidemiologic surveillance to better understand geographic clade distribution and the dynamics of vaccine versus wild-type strains. This has informed efforts to track the impact of vaccines and identify odd trends, like an increase in incidence reports during the COVID-19 pandemic. In order to lower the significant morbidity and financial costs associated with HZ, the epidemiology of the disease reflects a combination of host age and immune status, viral latency dynamics, changing varicella and HZ vaccination practices, and regional health-system capacity. These factors all support the need for ongoing, age-stratified surveillance and focused vaccination campaigns.11,13
Herpes zoster is caused by the varicella-zoster virus, which encodes a variety of virulence-linked genes that control immune evasion, viral replication, latency, and reactivation. At least 71 distinct ORFs are present in its linear double-stranded genome (~125 kb), many of which are conserved across herpesviruses and play crucial roles in pathogenesis. Important elements include glycoproteins that mediate viral entry, cell-to-cell transmission, and immune interaction, such as gB (ORF 31), gE (ORF 68), gH (ORF 37), gI (ORF 67), gK (ORF 5), gL (ORF 60), and others.14 gE, in particular, binds to an enzyme that breaks down insulin, which facilitates cell attachment and immune modulation. While immediate-early trans activators like ORF62 and IE63 (product of ORF63) drive viral gene expression, tegument and kinase proteins—ORF47 and ORF66—modulate the innate immune response.15 Notably, ORF63 is necessary for establishing latency in sensory ganglia, guaranteeing viral persistence despite its dispensability for initial neuro-invasion. Importantly, recent research has shown that ORF9 is a direct antagonist of the cytosolic DNA sensor cGAS, suppressing type I interferon responses and impairing innate immune detection.16–18 ORF9 is another essential tegument component that interacts with IE62 and gE to form a nexus for capsid envelopment and intracellular trafficking. While some structural players, like ORF10, ORF17, ORF21, and ORF40, aid in virion assembly, transcriptional activation, or capsid structure, structural actors like ORF54 encode the gateway protein for DNA packaging into nucleocapsids. Together, these virulence genes enable VZV to precisely balance immune evasion, lytic replication, stealth latency, and efficient reactivation—all of which are critical characteristics that underpin the pathophysiology of HZV and provide opportunities for the development of new antivirals and vaccines.19
2.3.1 glycoprotein E (gE)
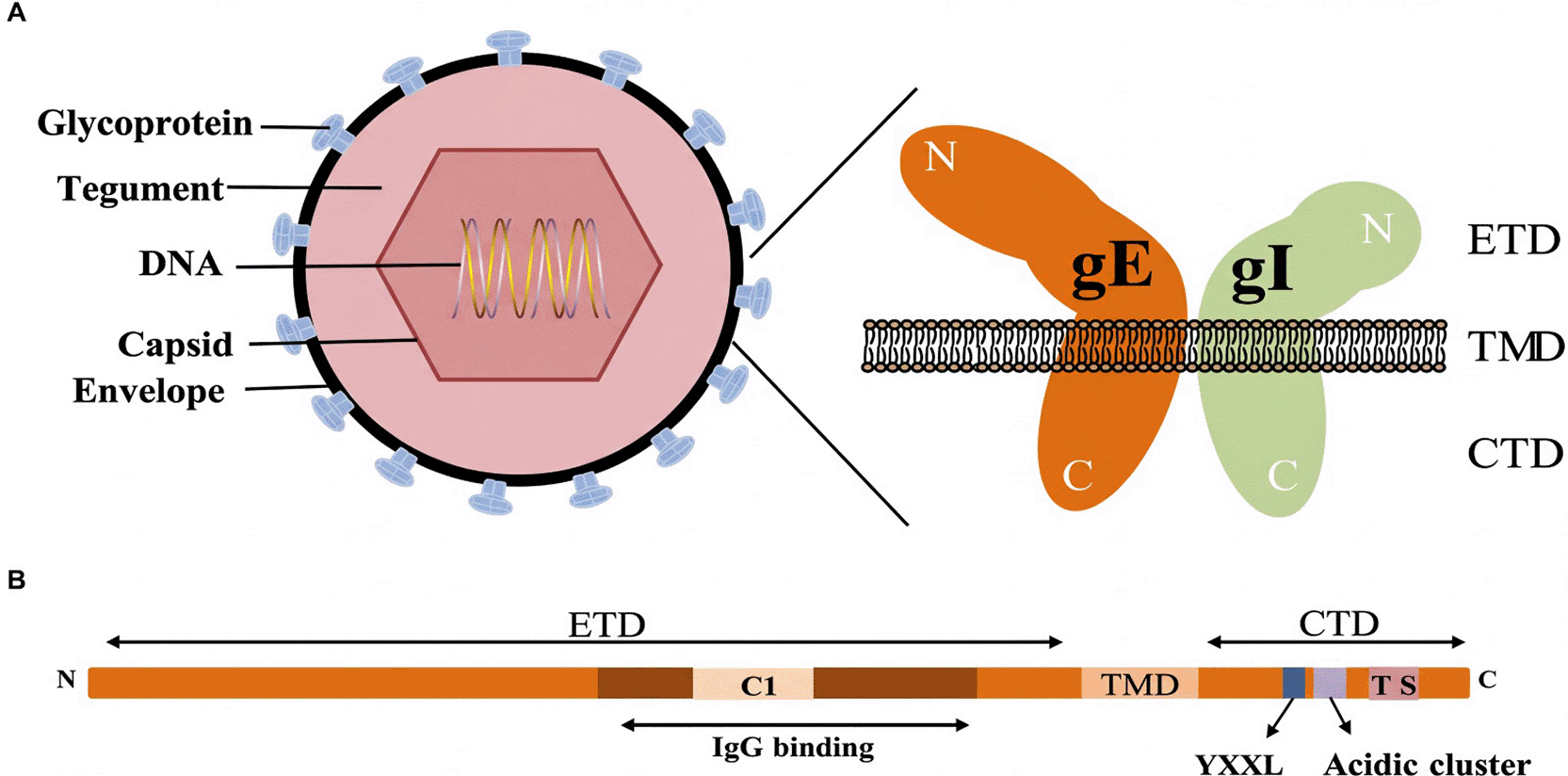
The varicella-zoster virus’s (VZV) glycoprotein E (gE) gene is a key virulence factor in the pathophysiology of HZV. This gene produces the most prevalent and vital viral glycoprotein, glycoprotein E, which is incorporated in the VZV lipid sheath as illustrated in (Figure 2).20,21 Functionally, gE coordinates three vital stages in the viral lifecycle: attachment to host cells, cell-to-cell transmission, and evading the host’s immune defense. Through binding with host cell receptors, the pathogenic capability of the virus is boosted by the facilitation of successful viral influx and viral dispersal in infected tissues. Glycoprotein I (gI) and gE also bind together to yield an elaborate complex enhancing viral cell-to-cell transmission with an impact on the ability to evade the host’s immune detection. Experiments illustrating gE gene deletions or mutant alleles lowering viral replicative virulence substantially prove the central role for gE in viral infectivity.22 Recent molecular studies remark on the functional need and strain conservation for the gE gene with a specific interest in its function as an antigen for future vaccination initiatives as also potential therapeutic targeting. In virtue of its multifunctional involvement in the regulation of immunity as also VZV pathogenesis, the glycoprotein E gene continues to represent the most critical virulence gene for the clinical manifestations and signs for herpes zoster, including the typical neuropathic pain and dermatomal rash.21,23–26 Due to its strong immunogenicity and pivotal role in viral pathogenesis, glycoprotein E is not only a good target but also the most clinically successful target for preventing herpes zoster at the moment.
2.3.2 Glycoprotein I (gI) gene
One major virulence factor for the Varicella-Zoster Virus behind the pathophysiology of herpes zoster (shingles) is glycoprotein I (gI). This 58–62 kDa glycoprotein is the protein produced by the open reading frame 67, and cell-to-cell transfer along with viral infectivity is fundamental for it. Specifically, human T cells as well as the skin are where cell-to-cell transfer as well as viral infectivity is fundamental for. Experiments indeed established that the gI protein is fundamental for viral reproduction along with in vivo pathogenicity by demonstrating that its deletion creates an infectious phenotype. In addition, heterodimer formation responsible for the increase in the capability for the virus to travel through the neural tissues depends on the communication between gI as well as glycoprotein E (gE).25,27 There indeed exists the potential for drug-readying this glycoprotein with drug agents’ courtesy of the establishment by current experiments that gI-specialized T cells maintain effector functions post-infection. In conclusion, the building of successful vaccines along with drug techniques for herpes zoster depends on the establishment of the mechanisms through which gI mirrors VZV pathogenicity.22,27,28 Indeed, the gI gene is a crucial virulence gene and a possible target for research on the herpes zoster virus, particularly in relation to comprehending immune evasion and viral spread. Though gI is more of a supplementary research target, gE is still the gold standard and is not the main target for vaccines or diagnostics.
2.3.3 ORF47 gene
One protein vital to the virulence as well as the pathogenesis of the Varicella-Zoster Virus, especially with herpes zoster (shingles), is the ORF47 gene. This protein is vital to the evasion of the virus by immunity responses in the host and is found in the distinctive small segment of the viral genome. From current studies, the ORF47 protein causes viral multiplication as well as survival in nervous tissues by binding with the different cell signal pathways in the host.26 Inhibition of apoptosis in cells infected with the virus has been demonstrated to enhance viral survival as well as virus re-activation out of latency. By the findings highlighted by newly conducted studies, the function played by ORF47 in the regulation of the immunological responses has been put across. This protein is also found to pose an effective therapeutic target. Writing effective vaccines as well as antiviral drugs for herpes zoster relies on the identification of the mechanism by which ORF47 increases the pathogenicity in VZV.29–31 Although it is not as clinically validated as gE, ORF47 is thought to be a good target gene for herpes zoster virus research due to its crucial role in replication and virulence. It holds greater promise for the development of antiviral medications than for routine diagnostics or vaccines.
2.3.4 Glycoprotein B (gB) gene
Glycoprotein B (gB), an integral virulence factor for the Varicella-Zoster Virus (VZV), is indispensable for the pathophysiology of herpes zoster (shingles). Since it ensures membrane fusion during infection, the gB protein, the gene product for the ORF31, has an integral role for the entry of the virus into the host cell. From the existing studies, gB has an obligatory function in the evasion of the immunity by regulating the host’s immunologic responses in addition to mediating viral attachment, fusion with the host cells.7 In particular, gB has the potential to suppress the efficacy of the host’s adaptive immunity by interfering with the presentation of the viral antigens to the CD8(^+) T cells. Also, alterations in the virulence and pathogenicity have been confirmed due to the mutations present in the gB gene, placing its significance in the pathogenicity for herpes zoster. In disruption or loss of the gB protein, VZV strains present decreased virulence largely, representing the indispensable need for the strong replication and virus spread. To identify the focused therapy as well as vaccines for herpes zoster therapy, information on the functional procedures for gB in VZV pathology is imperative.32–36 Although the gE gene is still the recommended target for vaccines due to its superior immunogenicity, the gB gene is a good target for herpes zoster virus research, particularly for molecular detection and antiviral strategies.
2.3.5 Glycoprotein H (gH) and Glycoprotein L (gL) genes
Essential heterodimeric glycoprotein complex H (gH; ORF37) and L (gL; ORF60), with gB, forms the primary fusion machinery of the VZV, mediating the entry of the virion and the syncytium-inducing cell-to-cell transmission aiding the pathogenesis of the skin neurons in herpes zoster. Since the gH/gL combination is necessary for the virus to replicate, neutralizing antibodies to gH highlight its function as a primary virulence factor by preventing VZV entrance and fusion. In terms of mechanism, gL acts as a chaperone that facilitates appropriate gH folding, maturation, and cell-surface distribution; mutational investigations reveal that disruptions in either partner lessen membrane fusion and polykaryon formation, which hinders dissemination. Control of gH density at the plasma membrane, including clathrin-dependent gH endocytosis, in infected tissues controls the degree of fusion and syncytia. Connecting the development of lesions and tissue damage to gH trafficking.37,38 Without a gD homolog, gH/gL expression and trafficking work with gB to carry out fusion in VZV, setting it apart from HSV and exposing unique receptor interactions that are currently being characterized. Domain-specific studies of gH further reveal cytoplasmic-tail and ectodomain contributions to fusion efficiency and skin tropism. The combination of tissue-tropism evidence, fusion regulation, antibody sensitivity, and genetic essentiality supports VZV gH/gL as important virulence genes with critical roles in primary varicella and reactivation illness.39 In order to study viral entry, fusion, and possible therapeutic interventions, the gH and gL genes are in fact good targets for the herpes zoster virus. However, rather than being the only primary targets, they are typically used in conjunction with gE for vaccine development or diagnostics.
2.3.6 Latency-associated genes
Varicella-zoster virus latency-associated genes, particularly the recently identified VZV latency transcript (VLT) and transcripts from open reading frame 63 (ORF63), are essential to the virus’s capacity to form, sustain, and eventually reactivate from neuronal latency. As a result, they indirectly contribute to the pathophysiology and virulence of herpes zoster. VLT is expressed antisense to ORF61 and appears to work with ORF63 to regulate latent state stability. During human ganglionic latency, expression is highly restricted and dominated by VLT and ORF63 RNAs.40 ORF63 transcripts are among the most abundant viral RNAs found in latently infected ganglia. Instead of acting as conventional lytic “toxins,” these latency-associated products modify neuronal transcriptional programs and have the potential to impact the threshold for reactivation, which is a critical determinant of salmonella incidence and severity.41 Further connecting latency-associated regulation with downstream virulence characteristics are tegument kinases expressed by ORF47 and ORF66, which take part in post-translational modification of viral and host proteins that impact neurotropism and replication competency following reactivation.42 A concept in which latency-associated genes function as modulators of viral persistence and reactivation propensity—two crucial virulence factors for HZV—is supported by the idea that these loci could be targeted for therapies aimed at reducing reactivation risk.43,44 Latency-associated genes are less useful as frontline targets for vaccines or immune-based therapies, but they are useful for comprehending VZV persistence and reactivation mechanisms and possibly for developing new antiviral strategies.
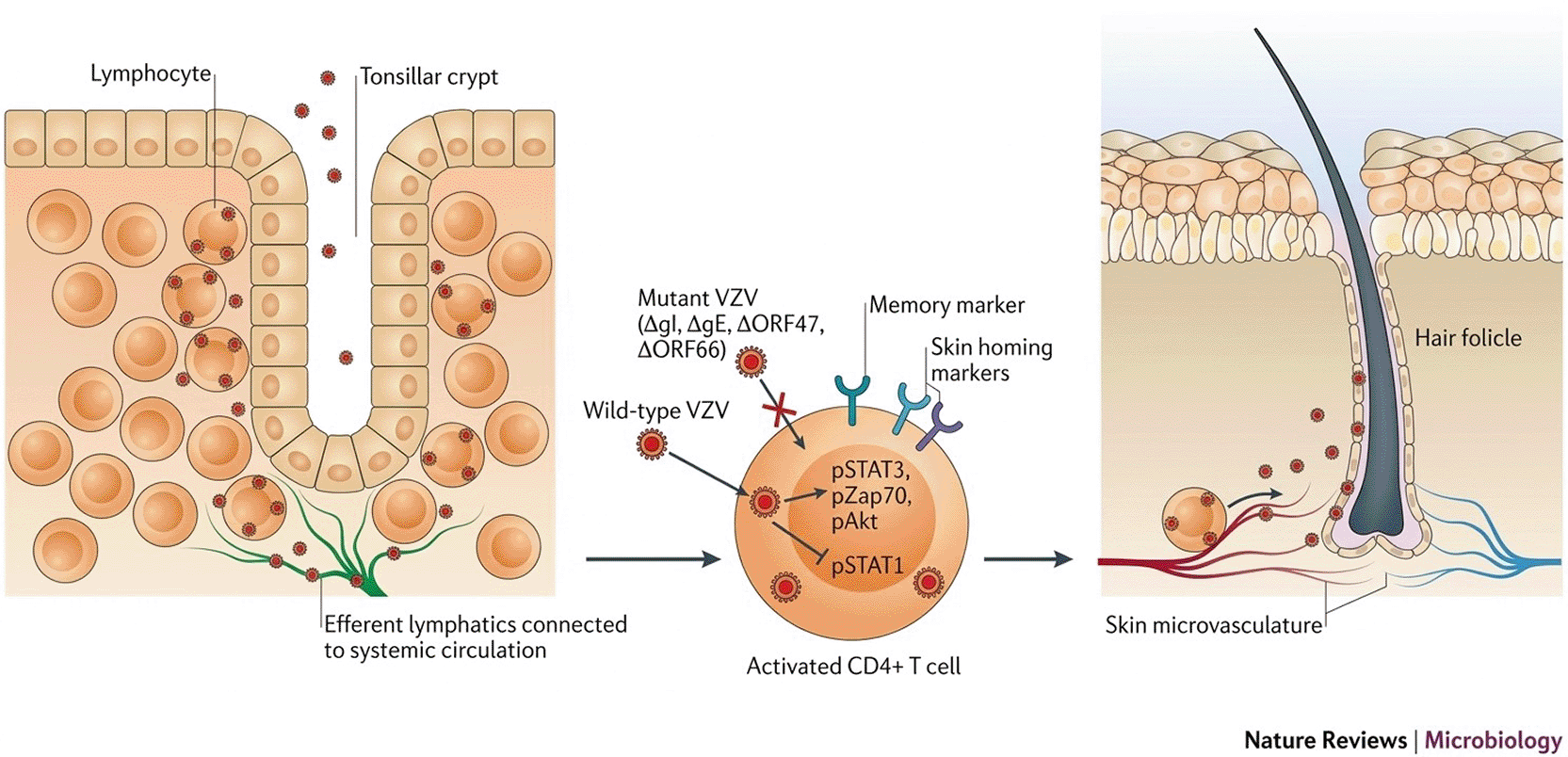
The pathogenesis of the varicella-zoster virus is a multi-stage process that starts with respiratory or close contact transmission of the cell-associated virus, then progresses to infection of mucosal and regional lymphoid tissues, cell-associated viremia mediated primarily by infected T lymphocytes, and widespread skin seeding where productive replication results in the characteristic varicella vesicular rash. This early systemic phase allows access to peripheral neurons that will be site as shown in (Figure 3).45 Following primary infection, VZV infiltrates sensory neurons and creates a deeply repressed latent state in the autonomic, trigeminal, and dorsal root ganglia. This state of latency is typified by exceptionally limited transcriptional activity, which is primarily dominated in humans by ORF63-associated RNAs and the varicella-zoster virus latency transcript (VLT), as well as by the epigenetic silencing of lytic promoters that collectively sustain transcriptional quiescence while retaining the possibility of reactivation.46 Both host factors (such as neuronal survival signals and chromatin-based repression) and virus-encoded regulators (such as latency-associated transcripts and proteins that can modulate viral transcription) are involved in the molecular control of this balance between latency and productive infection. Accordingly, changes in local signalizing or neuronal homeostasis would tip the balance in the direction of reactivation.40 Acute symptoms severity and long-term outcomes such as postherpetic neuralgia and, sometimes, involvement of the central nervous system are caused by the extent of viral reproduction, local inflammation, as well as neuronal destruction during this reactivation phase. Reactivation most frequently arises when cell-mediated immunity is down (most commonly due to ageing, immunosuppression, or wide-ranging stress), allowing afresh anterograde axonal transfer of infectious virus to the dermatome served by the harmed ganglion where localized viral reproduction causes the painful, unilateral, dermatomal herpes zoster eruption.41,42 Pathogenesis is the resulting emergent consequence of viral reproduction strategies, immigrant-evading behavior, as well as defense status for the host. Incidentally, VZV encodes numerous genes for products responsible for neurotropism as well as neuronal survivability, cell-to-cell dispersal through the dermis, as well as manipulation of innate as well as adaptive defense responses. Such understandings relating to mechanisms not only enlighten on the clinical scope of disease but also guide thwarting measures (e.g., vaccinating to enforce VZV-specific cellular defense) as well as therapeutic objectives focused on limiting reactivation as well as its aftermaths.47
Herpes zoster, which is caused by the latent varicella-zoster virus reactivating inside sensory ganglia, has very distinctive clinical symptoms that are strongly related to the virus’s underlying neurotropism and immunopathology. Prodromal symptoms, such as localized tingling, burning, or stabbing neuropathic pain, are typically the first to appear and may occur days before cutaneous lesions; they are indicative of viral multiplication and inflammation within the dorsal root ganglia and afflicted sensory nerves.49 Unilateral vesicular eruptions spread along a single dermatome, usually thoracic, cranial (especially trigeminal), or lumbar regions, are the hallmark dermatological finding. Over the course of seven to ten days, the rash progresses from erythematous macules to papules, vesicles, and finally crusted lesions.50 Although they are less frequent than in primary varicella, systemic symptoms including fever, malaise, exhaustion, and lymphadenopathy can nevertheless happen, particularly in people with impaired immune systems. One significant aspect of HZV is its correlation with neurogenic pain syndromes; postherpetic neuralgia (PHN), which is characterized by pain that lasts for weeks to months after the rash resolves, is the most common and incapacitating consequence, primarily in older people because of age-related declines in VZV-specific cellular immunity.51 Vesicle lesion sixty five years old female presented with HZ involved trigeminal nerve of ophthalmic division as illustrated in (Figure 4). Acute herpetic neuralgia is almost universal. When the ophthalmic branch of the trigeminal nerve is impacted, VZV reactivation can result in ophthalmic zoster, which can present as keratitis, uveitis, or vision-threatening consequences in addition to cutaneous and neurological involvement. Ramsay Hunt syndrome is a condition marked by vesicles in the auditory canal, ear discomfort, and facial paralysis that can also be brought on by involvement of the cranial nerve. Dissemination can manifest as encephalitis, pneumonitis, hepatitis, or broad vesicular eruptions in immunocompromised patients, indicating a lack of control over viral reproduction. Together, these symptoms are caused by direct viral cytopathic effects on the skin, neuroinflammation in sensory pathways, and host immunological responses. The severity and complications of HZV are highly determined by age and immune competence.52,53
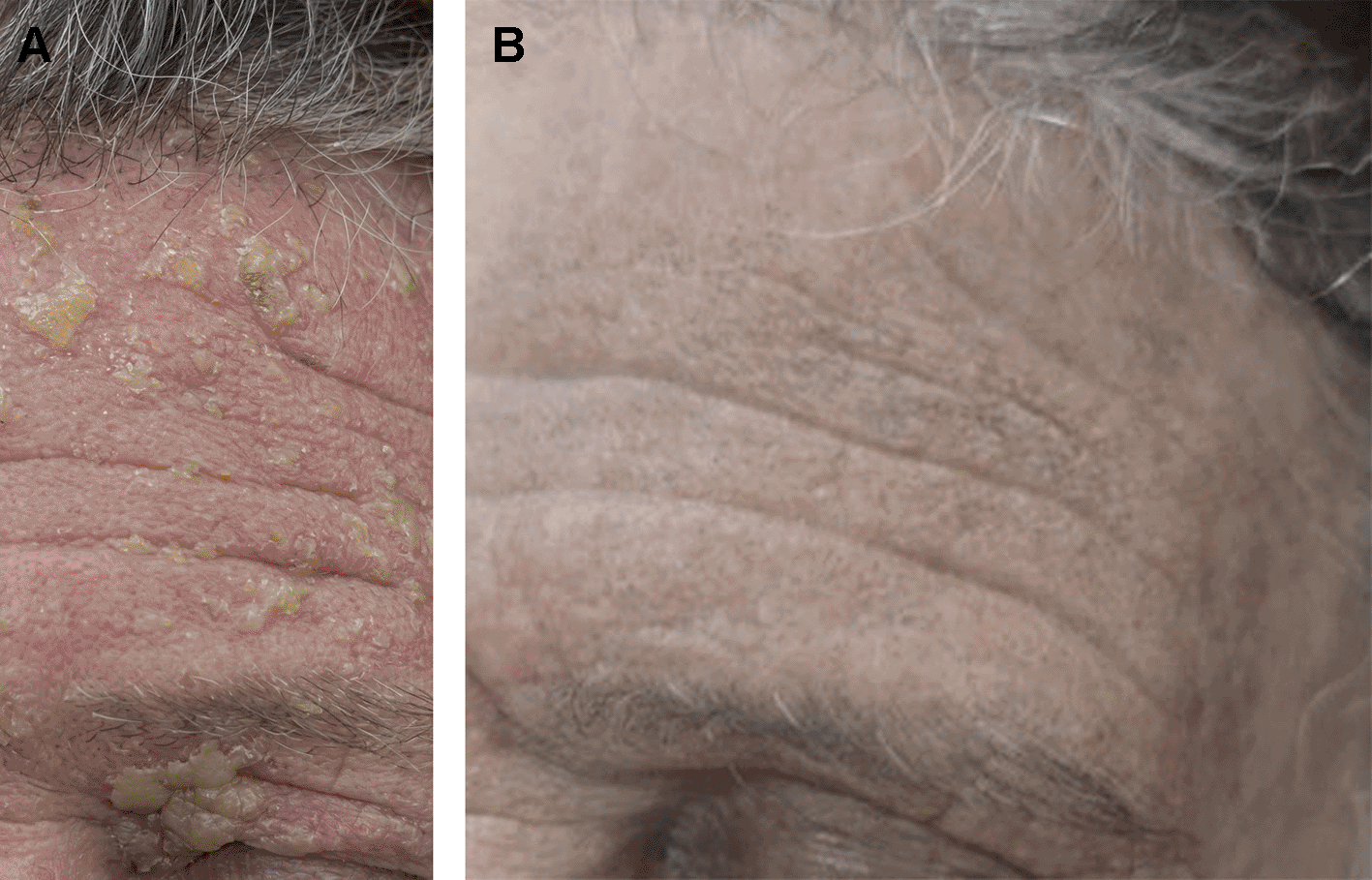
A. Before at 3 days of infection HZ While. B. After 3 months of healing with post inflammatory hyperpigmentation and association post herpetic neuralgia.
H. Zoster is a clinical entity caused by loss or weakness of immunity to varicella-zoster virus (VZV), an organized cooperation between early barrier-limiting innate defenses and adaptive, cell-mediated mechanisms responsible for long-term control as well as post reactivation controls. Innate sensors in infected epithelial and neuronal tissues—pattern recognition receptors expressed by keratinocytes, plasmacytoid dendritic cells, and macrophages—detect viral components early after exposure or during reactivation. They then trigger the production of type I interferon and proinflammatory cytokines, which limit local replication and prime adaptive immunity.54 VZV encodes several immune-modulatory proteins that suppress these responses, such as interferon signaling inhibitors and molecules that disrupt antigen presentation, which promote neuronal invasion and latency establishment. Conventional dendritic cells and natural killer (NK) cells also aid in early containment and cross-priming of T cells.55 The primary determinant of control is the adaptive immunological response, specifically VZV-specific cell-mediated immunity (CMI) mediated by CD4+ and CD8+ T lymphocytes: While CD8+ cytotoxic T cells restrict viral replication in infected tissues and probably monitor latently infected ganglia for reactivation events, CD4+ T cells aid in the generation of antibodies and the coordination of effector responses.56 Since antibodies by themselves cannot stop reactivation, humoral immunity (neutralizing antibodies) helps prevent cell-free spread and protect against primary varicella. This is why durable T-cell immunity is most strongly associated with lower incidence and severity of herpes zoster.57 VZV-specific CMI decreases both quantitatively and qualitatively with ageing (immune senescence) and in immunocompromised states, decreasing surveillance of latent virus and lowering the reactivation threshold. This epidemiologic and immunologic relationship explains why herpes zoster and its complications (like postherpetic neuralgia) are more common in older adults and immunocompromised patients.58 By using vaccination strategies that boost VZV-specific T-cell responses (like the recombinant zoster vaccine), restore protective CMI, reduce reactivation risk, and emphasize the significance of cellular immunity in preventing H. zoster, mechanistic immunology is translated into effective public health interventions.59
Clinical diagnosis of herpes zoster virus infection is primarily made by identifying the characteristic unilateral, dermatomal distribution of painful vesicular eruptions, which are often accompanied by prodromal neuropathic pain. However, laboratory confirmation is essential in atypical cases, patients with compromised immune systems, or presentations without rash (zoster sine herpete). PCR is the gold standard for laboratory diagnosis because it can quickly and thoroughly detect VZV DNA in tissue samples, blood, cerebrospinal fluid, or vesicle fluid. It is especially helpful for central nervous system (CNS) complications like encephalitis or meningitis.49 While serological testing (IgM and IgG antibodies) may help differentiate primary infection from prior exposure, its usefulness in acute zoster is limited due to the high prevalence of prior immunity. Direct immunofluorescence assays and viral culture are less sensitive and are rarely used in contemporary clinical practice. Although it is saved for patients that are difficult to diagnose, histopathological analysis of biopsy material in atypical cutaneous presentations reveals distinctive multinucleated giant cells and viral cytopathic alterations.60 Crucially, the diagnosis of zoster sine herpete depends on the intrathecal production of VZV-specific IgG antibodies in cerebrospinal fluid or the identification of VZV DNA by PCR.61 Real-time quantitative PCR is one example of how advances in molecular diagnostics have increased sensitivity, specificity, and speed, allowing for better handling of complex cases and distinction from other vesicular eruptions like herpes simplex virus infections. Overall, even though H. zoster is still primarily diagnosed clinically, ongoing advancements in molecular virology continue to improve the accuracy of diagnostic methods, and confirmatory laboratory testing employing nucleic acid amplification techniques is essential for atypical presentations and complications.62,63 Look to Table 1, contain herpes zoster virus vaccine strategies and diagnostic techniques.
Antiviral medication, pain management, and immune-prophylaxis are the mainstays of treatment and prevention for H. zoster infection; new developments have greatly improved outcomes for both immunocompetent and immunocompromised people. Early administration of antiviral medications, such as acyclovir, valacyclovir, and famciclovir, within 72 hours of the rash’s onset has been demonstrated to reduce acute pain, speed up lesion healing, shorten the time it takes for the virus to replicate, and reduce the risk of postherpetic neuralgia (PHN) by limiting neuronal damage.64 While corticosteroids may offer temporary pain relief, their limited long-term benefits make them unsuitable for routine use. To control acute neuropathic pain and prevent chronic pain syndromes, adjunctive therapy with analgesics, such as nonsteroidal anti-inflammatory drugs, opioids, gabapentinoids, or tricyclic antidepressants, is crucial.65 Vaccination is the mainstay of prevention methods since it has been shown to be the most successful way to lower the incidence and severity of HZV. The recombinant subunit vaccine (Shingrix®), which contains VZV glycoprotein E in conjunction with the AS01B adjuvant system, has largely supplanted the live attenuated zoster vaccine (Zostavax®), which showed moderate efficacy.66 It offers over 90% protection against HZV and PHN, with sustained efficacy across age groups, including adults over 70. Crucially, Shingrix provides greater protection for at-risk groups and is safe and highly immunogenic in immunocompromised patients, including those with solid organ transplants and hematologic malignancies. Development of vaccines against herpes zoster virus (HZV) has greatly transformed prevention of shingles and its complications, particularly in the elderly. The first effort was the live-attenuated vaccine (Zostavax®) licensed in 2006, which was based on the Oka/Merck strain of varicella-zoster virus. Zostavax reduced the incidence of herpes zoster by approximately 51% and post-herpetic neuralgia by 67%, but its effectiveness decreased very substantially between 5–8 years and was much less in individuals over the age of 70 years, limiting its long-term use. Advances in molecular virology and immunology led to the development of the recombinant subunit vaccine (Shingrix®), licensed by the FDA in 2017. Shingrix targets the highly immunogenic glycoprotein E (gE) together with the adjuvant system AS01B, inducing good and long-term CD4+ T-cell and humoral immunity. With a greater than 90% effectiveness across all ages, Shingrix has now come to be the gold standard for the prevention of HZV reactivation, curtailing disease burden, hospitalization, and healthcare costs significantly.67,68 As a result, current recommendations call for universal vaccination for adults over 50 and for younger immunocompromised people, with research into the best dosage and long-term immunity still ongoing. Combining early antiviral treatment with recombinant subunit immunization effectively manages the acute illness load and long-term consequences of herpes zoster while highlighting the importance of continuous immunological protection against VZV reactivation.69,70
Viral glycoproteins such as gE, gB, gH, and gL, and regulatory latency-associated genes, based on advances in molecular virology, are vital during viral entry, immune evasion, and persistence and hence are promising candidates for vaccine design. Current strategies, particularly subunit vaccines such as Shingrix®, have proven to be of high efficacy but with some challenges remaining. Research gaps are the lack of understanding of the molecular mechanisms of latency and reactivation, limited understanding of host–virus interactions in aging or immunocompromised hosts, and the need for improved correlates of long-term immunity. Additionally, while glycoprotein-based vaccines provoke robust immune responses, the duration and the protection breadth across populations with different genetics, comorbidities, and behaviors must be better measured. Future research highlights the potential for the conjoining of glycoprotein immunogens with latency-associated molecular targets, alongside new platforms such as mRNA, nanoparticle, or viral-vectored vaccines, to induce superior, more durable immunity. Closing these gaps will be essential to the creation of the next-generation herpes zoster vaccines that offer broader, more durable, and safer protection, reducing the global disease burden.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)