Keywords
Pediatric dilated cardiomyopathy, Prognosis, Risk stratification, Heart transplantation, Children, Echocardiography, Genetics, Trajectory
Pediatric dilated cardiomyopathy, Prognosis, Risk stratification, Heart transplantation, Children, Echocardiography, Genetics, Trajectory
Pediatric dilated cardiomyopathy (DCM) is a rare but very serious condition in which the heart chambers become enlarged and the heart’s pumping function is weakened.1,2 Usually, it’s the left ventricle (LV) that’s affected, but in some cases, both ventricles are dysfunctional.1 Modern pediatric classification systems mainly focus on the idea that adjusting for body size is an absolute must, and therefore LV dimensions and systolic indices are gen-erally understood with the help of age- and size-matched z-scores (e.g. LV end-diastolic dimension (LVEDD) z-score), together with clinical context and elimination of secondary causes.1 Despite advances in pediatric HF care, the condition of pediatric DCM still entails a major hazard of death or heart transplantation (HTx), especially quickly after diagnosis. Population-based registry anal-yses and national cohorts consistently demonstrate that events cluster in the first year, with a persistent but lower hazard thereafter. For example, a national population-based childhood DCM study reported that death or trans-plantation occurred in approximately a quarter of patients within 1 year, then about 1% per year thereafter, un-derscoring the “front-loaded” risk pattern.3,4 In the North American registry era, 1- and 5-year rates of death or transplantation were estimated to be roughly one-third and almost one-half, respectively, thus emphasizing how serious the condition has been over time.5 Meanwhile, it is equally feasible for patients to achieve recovery in a clinically significant subset of children. Besides, recov-ery has prognostic nuance itself. A huge registry data review of presumably idiopathic pediatric DCM with re-peated echocardiographic data confirmed that about 20% of patients normalized their LV size and function within a 2-year period. On the other hand, a smaller number of these patients later worsened and died or had to be transplanted, thus the term “normalization” may imply ei-ther recovery or remission depending on the underlying biology and duration of the follow-up.6 The consid-erable variations in disease patterns make it difficult to predict the course of the disease and to make the right clinical decisions. Families need to be given realistic ex-pectations; at the same time, clinicians have to decide on the appropriate level of monitoring, when to refer patients to advanced HF/HTx centers, if and when to choose me-chanical circulatory support, and whether or not mutant arrhythmia surveillance/devices should be considered in selected patients. Prognostic factors thus have a triple role: (i) to estimate outcome probabilities (death/HTx vs recovery), (ii) to lead risk-stratified management (in-cluding escalation pathways), and (iii) to identify patients for clinical trials and mechanistic studies. Moreover, the prognostic evidence is largely influenced by the context of care. A model developed in a wealthy environment where the patient has access to biomarkers, serial imaging, ge-netic testing, ventricular assist devices (VADs), and trans-plantation is unlikely to be applicable in a setting with a late diagnosis, limited capacity for follow-up, and lack of advanced therapies. Recent articles from7,2 showcase the types of obstacles at the system-level and the differ-ent results that may have been found in well-established registries, thus emphasizing the need for prognostic inter-pretation that is aware of equity issues.8 In this review, we intend to: (1) provide a summary of the main prognos-tic factors in pediatric DCM with the respective levels of evidence for each; (2) group prognostic factors according to clinical significance and effect on outcomes; (3) depict an integrated model that connects symptoms, risk levels, and outcomes (giving a concept map as well); and (4) highlight the areas of research that are missing and there-fore hinder the availability of generic, clinical pediatric risk prediction.
This paper is a narrative review that aims to provide a clin-ical interpretation and manuscript-ready synthesis, rather than a systematic and exhaustive listing of the cases. Lit-erature priority was agreed on the basis of three princi-ples: (i) focus on primary research in pediatric DCM (reg-istries, national population-based cohorts, multicenter ob-servational studies), (ii) acceptance of recent systematic reviews/meta-analyses on pediatric prognostic imaging factors, and (iii) use of major consensus/guideline state-ments which are relevant to pediatric HF care, diagnos-tics, implantable electronic devices, and cardiomyopa-thy evaluation. We compiled evidence from various do-mains: (a) demographics/etiology, (b) clinical HF sever-ity and trajectory, (c) electrocardiography/arrhythmias, (d) echocardiography and CMR, (e) biomarkers and func-tional capacity, (f ) genetics and family history, and (g) context-of-care modifiers. Death, heart transplantation, VAD implantation as an advanced HF endpoint, recov-ery/normalization of LV size and function, and sudden cardiac death (SCD) or arrhythmic events were the main outcomes of interest where pediatric DCM, specific data were available.
We reconstruct the principal prognostic factors in pedi- atric DCM on the basis of recent high-quality evidence and consequently we arrange them categorically.
A. Clinical and Demographic Prognostic Fac-tors
Age at diagnosis has always been an important prognos-tic factor for pediatric DCM patients from a clinical per-spective.5 Later childhood/adolescence at presenta-tion usually exhibits worse transplant-free survival than younger children among several cohorts.2,3 Older age at diagnosis in the PCMR (for instance, above 6 years) was still a risk factor for death or heart transplantation independently of the other confounding factors.2 Sev-eral series have considered young age as a positive prog-nostic factor possibly due to a higher proportion of re-versible conditions at early ages (such as myocarditis) and an ability to recuperate for those who survive the initial very risky period.5,2 Nevertheless, infant-onset DCM (< 1 year) is not always protective; some groups find that infants have a higher short-term mortality rate, which is probably due to the disease progressing so quickly that there is no time to get advanced support.4 This is in line with the idea that age, risk relationship is non-linear, with the highest risk being at the extremes of age (very early infancy and older childhood) rather than a simple linear effect.4 From a clinical perspective, adolescent-onset DCM is a case for a higher level of risk alert, whereas the age factor should be considered in combination with etiology and severity at presentation.2,4,9
In pediatric DCM, etiology serves as the main “risk con-text” since it determines how severity markers (EF, LV dila-tion, MR, and biomarkers) are to be interpreted.2 Data from the registry era reveal a large variation in the out-come depending on the cause, with significantly better scenarios in the categories of potentially reversible versus primary/genetic phenotypes.2 Different studies show that myocarditis-associated DCM is more likely to recover function and have fewer death/HTx events than idio-pathic DCM, which is in line with an acute inflammatory injury that may be facilitated by early support.2,10 By contrast, idiopathic DCM, which is largely accounted for genetic cardiomyopathy cases in modern cohorts, has been associated with lower transplant-free survival and a more deteriorating progression of the disease.2,11 Familial/genetic disease could be at a higher risk in some cases, and the family history can be a convenient bedside indicator for increased monitoring; for instance, a single-center cohort study found family history as a risk fac-tor in time-to-event analysis.12 Neuromuscular DCM (e.g., dystrophinopathies) is also typically linked to worse prognosis, which is influenced by the presence of sys-temic comorbidities and, in some cases, limited advanced-therapy candidacy.2 Importantly, new data have con-nected the words “etiology” and “genotype”: in a study involving children who had their myocarditis confirmed through biopsy and showed a DCM phenotype, it was found that pathogenic/likely pathogenic cardiomyopathy variants were highly present and that event-free survival was significantly lower in those that had such variants, thus indicating that myocarditis may reveal an inherited substrate.10,13 Therefore, the etiologic classification should be clear when talking prognostics, as it not only changes the baseline risk but also how genetic testing can further refine the risk even within presentations labeled as myocarditis.2,10,13
Severity of HF at diagnosis is strongly indicative of a near-term prognosis in pediatric DCM. Children manifesting advanced symptoms (Ross/NYHA III, IV) are found to be at a notably greater risk of death/HTx as compared to those with less severe presentations.14 In a signifi-cant group of children, the researchers created a nomo-gram and a simple bedside score. They found that an advanced functional class not just predicted the outcome but was also the factor with the greatest weight, which is in agreement with the idea that severe heart failure is a highly indicative marker of immediate danger.14 Markers of critical illness, such as admission to the ICU, being on inotropes for a longer period, and the need for MCS, are mainly seen in children very close to cardiogenic shock and thus, these markers are highly correlated with early death or very quick progression to HTx.15 Besides baseline visual acuity being a starting point, prognosis is not static: analysis of registry data shows that the greatest risk of death or heart transplant is right after the diagno-sis, and those who stabilize continue to have an improv-ing conditional prognosis with recovery patterns varying according to the cause and treatment response.2,16 On the clinical side, when a patient is in a critical condi-tion (for example, low blood pressure, which keeps get-ting worse, needing increasing doses of vasoactive drugs, ventilatory support, or consideration of ECMO/MCS), it is advisable to seek the help of advanced HF/transplant services immediately.15,14 Severity scores are also useful; for instance, elevated NYU-PHFI scores have been linked with greater chances of death/HTx in pediatric DCM populations.17
Arrhythmias in pediatric DCM patients can be complica-tions as well as signs of the existence of a more malignant myocardial substrate. Although atrial arrhythmias and conduction disease usually are signs of an advanced or arrhythmogenic phenotype, sustained VT and advanced AV block are directly relevant to the risk of sudden death.5 Previously, reviews showed inconsistent relation-ships between heart rhythm disorders and results, prob-ably because of different definitions and small numbers of events in cohorts.5 Current evidence from the reg-istry era confirms that physicians should classify as “high-concern” cardiac arrest or life-threatening ventricular ar-rhythmias and severe conduction abnormalities on ECG, notably when there is also severe LV dysfunction or the presence of adverse remodeling.18 Sudden cardiac death (SCD) is a smaller, but still significant, part of the total mortality in DCM (3% at 5 years). Hence, prag-matic stratification tools have been suggested that can be used to identify children who might be monitored more closely for their heart rhythm or even considered for de-vice therapy.18 For example, in one model, it was demonstrated that the majority of SCD events occurred in children who at one point fulfilled the triad criteria (severe LV dilation, age below a certain threshold, and LV wall thinning), which is in line with a thin-walled, heavily remodeled ventricle that is vulnerable to malig-nant arrhythmias.18 ECG phenotypes might provide additional prognostic information that is independent of other factors: lowvoltage QRS was independently asso-ciated with death/HTx in a contemporary pediatric risk score,14 probably indicating diffuse myocardial disease or fibrosis burden.14 In addition, conduction disease may indicate the presence of high-risk genotypes; LMNA-related DCM is typically accompanied by conduction ab-normalities and malignant ventricular arrhythmias, which justifies rhythm surveillance and escalation planning from a very early stage.19 A similar example would be SCN5A-related phenotypes, which may present a mixture of conduction disorder/arrhythmias, together with struc-tural DCM, thus emphasizing the idea that “electrical red flags” might be an indicator of genotype-related risk.20 In a real-world scenario, a patient with sustained VT, a high number of ectopy/NSVT together with adverse re-modeling, or major conduction disease should be consid-ered for a more intensive follow-up and the involvement of an electrophysiologist, while decisions about the device should be individualized.18,19,20
The knowledge of cardiomyopathy or sudden cardiac death in a family is medically very significant as it can be used as a proxy for the presence of hereditary dis-eases. Therefore, it is common that this evidence leads to early genetic testing and cascade screening.21 On the other hand, the majority of studies show that family his-tory alone has no clear effect on the outcomes of pediatric DCM. The variations in definition, referral patterns, and genetic testing intensity among cohorts might have con-tributed to the different evidence obtained.22 A single-center cohort found a positive family history of DCM to be significantly associated with mortality in Cox regression.12 On the other hand, a registry-based comparison of familial DCM versus idiopathic DCM in children revealed a broadly similar transplant-free survival, and the family history of sudden death was not an independent predic-tor of death/HTx.22 Similarly, in the context of SCD-centric modeling, family history has not always been ob-served as a standalone predictor.18 Thus, family his-tory should ideally be handled less as a statistical risk-enhancing factor and more as a very effective prompt for obtaining a more comprehensive etiologic understanding, especially genetic testing and rhythm monitoring, since the actual risk is frequently determined by the genotype-associated malignant phenotypes rather than by family history itself.21,22
While the occurrence rates differ between sexes, sex, however, has not ultimately been identified as a stable independent predictor of death/HTx when the pheno-type and severity are taken into account.2,22 Un-der competing-risk models, male sex has failed to demon-strate a strong correlation with the combined outcome of death or transplantation, thereby implying that the dif-ference seen is usually a result of the severity, etiologic mix, or care pathways factors.22 For example, sex-related disparities in advanced-therapy treatments rather than in the biology of intrinsic DCM could be more pro-nounced; transplant-era analyses, for instance, have doc-umented differences in pediatric waitlist survival by sex while the transplantation rates were the same.23 In comparison, growth and nutritional status are indicative of the overall whole-child severity. Stunting (low height-for-age z-score) has been linked in PCMR studies to a higher risk of death (even more than the risk of trans-plantation), which is in line with the understanding that growth failure reflects the chronic burden of heart fail-ure and the child’s susceptibility to decompensation.5 From a clinical standpoint, failure to thrive or malnutri-tion can be considered effective warning signs that ne-cessitate increased monitoring and supportive treatment when combined with adverse cardiac indicators.2,5
Resource setting has a major impact on the outcomes ob-served in pediatric DCM, mainly through access to ad-vanced HF therapies such as HTx and durable VAD sup-port.24 In LMICs, these options may be limited, and outcomes often reflect what can be achieved with med-ical therapy and supportive care alone; a prospective study conducted in Khartoum state (Sudan) has shown very high mortality rates, thus emphasizing the suffer-ing of the very ill patients without the availability of scal-ing up treatments.25 Other LMIC cohorts also report similar high event rates and persistent severe dysfunc-tion, which is in line with delayed presentation and lim-ited longitudinal HF infrastructures that exacerbate early risk.26 LMIC studies also uncover modifiable factors. For instance, an Ethiopian decade-experience series first linked malnutrition to bad outcomes and then enalapril use to better outcomes, implying that basic HF optimiza-tion in these patients can still bring significant improve-ments even when HTx is not available.27 On the other hand, in transplant-capable systems, HTx and durable support have the potential to change the paths of the pa-tients from death to survival with advanced intervention, thereby making the use of the composite endpoint “death or HTx” complicated for comparisons.24 Experience in the United States shows that using durable VADs as a bridge to transplant for a longer period can result in greater success of bridging to transplantation when com-pared with the use of extracorporeal support. This is an example of how the healthcare system’s ability to man-age a case can change what was initially a fatal course into a survival after transplantation.28 Through the years, registry studies have also revealed enhanced sur-vival without the need for a transplant, indicating that HF treatment improvements have been made in general and not just in the areas of transplant volume.7 Hence, prognostic understanding must deliberately recognize re-source context; a given clinical severity, therefore, may result in different actual risks based on whether and how quickly advanced therapies are available.24,25 The major clinical and demographic predictors, together with representative supporting evidence, are summarized in Table 1.
B. Echocardiographic and Imaging Predictors
Since DCM basically is ventricular dilatation and dysfunc-tion, one should not be surprised that quantitation of the cardiac size and function by various imaging techniques have very powerful prognostic significance. Echocardio-graphy is the first interventional tool and a number of studies have tested ejection fraction, fractional shortening and ventricular dimensions as predictors of outcome. Re-cently, different advanced imaging parameters like strain from speckle-tracking echocardiography or fibrosis detec-tion by cardiac MRI have been investigated. A systematic review and meta-analysis focusing on childhood imaging risk factors in DCM by Street-de-Palma et al. (2025) con-firmed EF, LV size, and MR to be the top three most consistently associated parameters (refer to Table 2 for pooled effect estimates).8
LVEF is still one of the most consistent and reliable imag-ing markers for pediatric DCM. It has been able con-sistently to distinguish between higher- and lower-risk groups not only in individual observational cohorts but also pooled analyses.5,8 Fractional shortening (FS), which is commonly used in pediatrics, gives a supplemental prognostic indicator; initial FS (and its standardized z-score) has been associated with death/HTx risk, and, clin-ically important, serial absence of FS improvement identi-fies children with continuously high risk even when base-line values are similar ( Table 2).29 In fact, EF/FS can be regarded as severity anchors that provide the bound-aries for the rest of the prognostic assessment and thus should not be seen as standalone “yes/no” predictors.5
Besides it being merely a baseline severity, the early re-modeling trajectory typically determines the fate. Se-rial PCMR analyses reveal that, at the point of one year, the majority of children who are alive without a trans-plant have their LV systolic function and chamber size im-proved. However, the cases where the function remains impaired or the dilation progresses in the first few months of the disease are marked as a high-risk pathway.30 This notion of “dynamic risk” is in line with shifting from a single time-point interpretation (baseline EF/FS) to-wards a trajectory-based interpretation (response vs non-response) in consultation with families and making deci-sions on the timing of escalation.2,16,30 Besides, it is necessary to integrate trajectory with other domains; for instance, a longitudinal study might find that imaging measures (such as GLS) were significant predictors in the univariate analyses, however, NT-proBNP was identified as the strongest independent predictor in the multivari-ate analysis, meaning that remodeling parameters usu-ally reflect the same heart failure biological process that biomarkers detect (and not that they are substitutes for biomarkers).31
LV dilation that is usually measured as LVEDD z-score is the second main imaging predictor of pediatric DCM and has been consistently negatively associated in pooled ev-idence ( Table 2).8 Longitudinal modeling highlights that changes over time may even represent a larger risk factor than baseline measurements. LV size at baseline is important, but if the size keeps getting worse in se-rial measurements (particularly enlargement of systolic dimensions), it usually reflects an unfavorable remodel-ing process and increased risk of events.32 At the bed-side stratification, simple threshold-based methods have been suggested to help in identifying high-risk pheno-types (e.g., CART-derived cutoffs combining LV size and EF). These methods confirm that the combination of “very large LV + severe dysfunction” is widely recognized as the most dangerous pattern across datasets.33
Functional mitral regurgitation is common in children with idiopathic DCM and is a direct consequence of ge-ometric changes (annular dilation and papillary muscle displacement) occurring together with a deterioration of hemodynamics. Whereas some of the older studies dis-agreed, a collection of up-to-date evidence now indi-cates that moderate-to-severe MR is an important nega-tive prognostic marker ( Table 2).8 Actually, this idea has also made its way into prognosis tools; e.g., latest pe-diatric risk models use MR grade as a significant compo-nent to determine the risk of death/HTx, which is in line with MR being both a marker and a booster of remodel-ing (volume overload and reduced forward output).14 From a clinical standpoint, the level of MR provides the most valuable information when it is combined with LV size and systolic function, and also when it is monitored over time as the remodeling changes continue.8,14
Even though the strongest predictors in prognostic mod-els are typically those related to the LV, the presence of RV dysfunction frequently signifies a progressed biventricu-lar disease and can challenge the management as well as the planning of advanced therapies. Serial studies on chil-dren have revealed that intact RV function (for instance, greater TAPSE or RV fractional area change) is linked to a lower risk, which is in line with the clinical view of RV reserve being protective ( Table 2).32 Hence, measures of RV function should mainly be considered as a potential additional source of risk information, especially when one is thinking about the timing and suitability for therapy es-calation.32
Speckle-tracking GLS is a technique that analyses myocar-dial deformation, which means that it is able to uncover dysfunctions that EF cannot detect. In children, GLS has been a significant prognostic factor in univariate analyses of repeated-measures cohorts, but its incremental inde-pendence may be compromised when powerful biomark-ers of HF (especially NT-proBNP) are added to the model, thus thereby partially sharing the signals of global HF severity.31 Anyway, GLS remains an absolutely valid clinical instrument for profiling phenotypes and is highly sensitive for the detection of subtle systolic impairment; in the case, control pediatric data, GLS was found to be greatly impaired in DCM compared to normal controls, thus depicting the magnitude of deformation loss in the disease condition.34 Today, GLS mainly serves as a secondary marker, for instance, in cases where EF is bor-derline or when the initial phases of the disease are being tracked, while waiting for a large pediatric dataset to help quantify the additional prognostic benefit over EF/LV size and biomarkers.31,34
CMR offers additional tissue characterization, such as LGE which is used as a fingerprint of fibrosis/scar. LGE is a very well-known negative marker in adult DCM but prog-nostic evidence in the pediatric population is still scarce. According to the 2025 meta-analysis, only a few pediatric studies on LGE were found and a statistically strong cor-relation with outcomes was not shown, thus highlighting the limited power and heterogeneity of the present pe-diatric CMR literature ( Table 2).8 Therefore, CMR is typically very helpful in children mainly for clarifying the cause of a disease (for example, patterns of myocarditis or infiltrative diseases) rather than for final risk assessment, however, in practice, widespread fibrosis is still generally considered worrying.8
Several additional echo measures, particularly diastolic indices, show emerging albeit inconsistent evidence in pe-diatrics. In longitudinal prediction studies, certain dias-tolic surrogates and their serial changes were linked to the lowering of risk, thus they supported the idea that di-astolic remodeling patterns could, in certain phenotypes, contain clinically relevant information.32 Smaller ret-rospective analyses have also indicated a strong correla-tion of very low transmittal E-wave velocity with mor-tality, possibly due to severely reduced forward flow or advanced dysfunction in certain children.12 Older global size surrogates (e.g. CTR on chest radiography) in essence are a mirror of the echo derived cardiomegaly and have been reported as adverse in some earlier contexts, however, echo has essentially taken over CTR for current risk quantification.9 Pulmonary hypertension (PH) de-tected by echocardiography (e.g., high estimated RV sys-tolic pressure from TR jet) is an extra prognostic factor as it mostly indicates that the LV filling pressures have been elevated for a long time and the disease is at an advanced stage. Among the pediatric DCM group reported by Wang et al., PH (defined by PASP ≥30 mmHg) was a frequently observed clinical condition and it was even more numeri-cally common among non-survivors, However, PH did not turn out to be an independent predictor when multivari-able modeling was done. This is just one example of how PH might be a severity correlate instead of an indepen-dent signal in different datasets.35 From a clinical per-spective, one of the key factors that can lead to the accel-eration of referral and escalation planning (including ad-vanced HF/transplant assessment) is the demonstration of persistently elevated pulmonary pressures despite op-timized therapy, as this could potentially limit the options and also increase the risk during the peri-transplant pe-riod.36
C. Biomarkers and Neurohormonal Indicators
Circulating biomarkers add objective, repeatable signals of hemodynamic stress, neurohormonal activation, and systemic response that complement clinical and imag-ing assessment in pediatric DCM.31,37 Natriuretic peptides are the most extensively validated for progno-sis among the various laboratory biomarkers available, whereas inflammatory indices that are derived from CBC and abnormalities in routine chemistry are probably best regarded as adjuncts, with very limited pediatric evidence at present ( Table 3).12,31,37
1) NT-proBNP/BNP
| Factor | Association with outcome | Evidence |
|---|---|---|
| Older age at diagnosis | Worse prognosis (higher risk of death/HTx). Younger age tends to be protective (though neonatal‐onset can have high early mortality). | – PCMR: older children had higher 5‐year risk of death/HTx.2 |
| – Alvarez 2007 review: younger age was associated with better survival in multiple studies.5 | ||
| Etiology: myocarditis | Better prognosis (often transient disease). Higher likelihood of recovery and lower transplant rate compared to idiopathic/genetic DCM. | – PCMR: 5‐year event‐free survival about 73% in myocarditis vs about 47% in idiopathic.2 |
| – Multiple cohorts: myocarditis diagnosis predicts improved LV function and survival vs primary DCM.2,10 | ||
| Etiology: idiopathic/genetic | Worse prognosis. Progressive disease course, low chance of recovery. Familial cases are often higher risk (depends on genotype). | – PCMR: idiopathic DCM had the poorest outcomes (5‐year event‐free about 47%).2 |
| – Arici 2025: family history of DCM associated with about a 3‐fold higher mortality risk in one cohort.12 | ||
| Etiology: neuromuscular | Very poor prognosis. Often progressive despite therapy; systemic disease limits transplant options in some cases. | – PCMR: 5‐year event‐free about 52% in neuro‐muscular DCM (among worst outcomes).2 |
| Heart failure class III–IV | Worse prognosis. Severe CHF symptoms at presentation predict early death or transplant. | – Xu 2025: NYHA/Ross class III–IV was an independent predictor (score weight 10 points).14 |
| – Gashaw 2024 (Ethiopia): class IV HF at diagnosis associated with higher mortality.27 | ||
| Need for ICU/inotropes/MCS | Very high‐risk prognosis. Indicates cardiogenic shock; often leads to urgent transplant or death if not rapidly supported. | – Xu 2025: need for vasoactive infusion predictive of death/HTx (included in risk model).14 |
| – Dutch cohort: about 41% required ICU & inotropes at presentation; this subgroup had high early event rates.15 | ||
| Arrhythmia (VT, VF, etc.) | Worse prognosis. Associated with sudden death risk and indicates advanced or arrhythmogenic myocardial disease. | – Pahl 2012: children on antiarrhythmic medication (for arrhythmia) had a 3‐fold higher risk of SCD.18 Five‐year SCD incidence about 2–3%; risk higher if severe LV dilation and wall thinning.18 |
| ECG low voltage or heart block | Worse prognosis. Low‐voltage QRS or significant conduction block may signal diffuse disease or high‐risk genotype. | – Xu 2025: low‐voltage ECG was an independent predictor (OR about 4.25) in a risk score.14 |
| – LMNA mutations: often present with AV block and carry high sudden‐death risk.19 | ||
| Family history of DCM/SCD | Suggests genetic DCM (heritable substrate). By itself, not consistently predictive, but often associated with malignant genotypes. | – Arici 2025: family history of DCM associated with about a 3‐fold higher mortality risk in one cohort.12 |
| – Rusconi 2017 (PCMR): no significant outcome difference familial vs idiopathic (after adjustment).22 | ||
| Female sex | Not a consistent independent factor (after adjustment). Some reports note differences in treatment pathways (e.g., waitlist outcomes). | – Most studies: no significant sex difference in survival after accounting for other factors.22 |
| – Bhimani 2022: females had worse waitlist survival than males despite similar acuity.23 | ||
| Poor growth/nutritional status | Worse prognosis. Indicates chronic HF impact (catabolic state); correlates with higher risk of death (vs transplant). | – PCMR: lower height‐for‐age z associated with in‐creased mortality (vs transplant).5 |
| – Gashaw 2024: severe malnutrition at presentation linked to higher mortality (p=0.017).27 |
| Imaging factor | Adverse prognostic association | Evidence |
|---|---|---|
| Left ventricular ejection fraction (EF) | Lower EF = worse prognosis. EF is a strong predictor of death/HTx risk. Each 10% drop in EF significantly increases risk. | – Meta‐analysis: HR about 0.80 per +10% EF (p=0.04), i.e., 20% risk reduction per 10% EF improvement.8 |
| – Multiple cohorts: non‐survivors have significantly lower EF at diagnosis than survivors.5 | ||
| LV end‐diastolic dimension (LVEDD) | Larger LV size = worse prognosis. High LVEDD z‐score is associated with higher mortality/HTx risk. | – Meta‐analysis: HR about 1.43 per +1 increase in LVEDD z (95% CI 1.13–1.81).8 |
| – PCMR: extremely large LV (e.g., LVEDD z ≥7.7) identified highest‐risk subset (poor 1‐year outcomes).33 | ||
| Severe mitral regurgitation (MR) | Severe MR = worse prognosis. Indicates advanced remodeling; linked to higher risk of HF death or transplant. | – Meta‐analysis: OR about 5.1 for severe MR vs none/mild (p=0.004).8 |
| – Xu 2025: moderate–severe MR was an independent predictor in a risk model (assigned 2.5 points).14 | ||
| Lack of early LV improvement | Failure of EF and dilation to improve in first 6–12 months = worse prognosis (persistent severe dysfunction/dilation signals high risk). | – PCMR: patients whose LVEDD increased in the first year had higher subsequent risk (HR about 1.45 per +1 z).30 |
| – van der Meulen 2021: those who died/HTx had minimal EF improvement vs survivors who showed significant improvement by 6–12 months.31 | ||
| Right ventricular dysfunction | RV failure = worse prognosis. Biventricular failure portends high mortality and can complicate transplant candidacy. | – Ishii 2021: lower RV FAC and TAPSE were associated with higher risk (worse survival).32 |
| Global longitudinal strain (GLS) | Worse (less negative) GLS = worse prognosis. GLS detects subtle systolic dysfunction; more abnormal strain correlates with higher risk, though it tracks with EF. | – Case–control: DCM patients had markedly impaired GLS (about –11%) vs controls (about –20%).34 |
| – van der Meulen 2021: GLS was worse in those who reached the endpoint vs event‐free patients.31 | ||
| Cardiac MRI fibrosis (LGE) | Fibrosis on MRI: unclear prognostic impact in pediatrics so far (insufficient data). In adults, LGE is high‐risk (scar predicts arrhythmia and death), but pediatric studies have not yet proven this. | – Meta‐analysis: small pediatric studies showed no statistically clear link between LGE and outcomes.8 |
| Diastolic function (e.g., E‐wave, E/e') | Very restrictive filling or low transmitral flow = likely worse prognosis (indicates poor cardiac output or high filling pressures). | – Arici 2025: mean mitral E velocity was significantly lower in children who died (0.78 ± 0.18 m/s) vs survivors (0.94 ± 0.22 m/s), p=0.031.12 |
| Pulmonary hypertension (PH) | Likely worse prognosis if present. Signifies long‐standing high LV filling pressures and may limit transplant options. | – Clinical consensus is that significant PH in DCM (e.g., TR jet >30 mmHg) indicates advanced disease and can increase risk.35,36 |
| Cardiothoracic ratio on CXR | Very large heart on chest X‐ray correlates with poor outcome (reflects severe cardiomegaly). | – Sudan cohort: massive cardiomegaly on X‐ray was common in non‐survivors.25 |
| Biomarker | What high levels indicate & prognostic impact | Evidence |
|---|---|---|
| NT‐proBNP | Reflects ventricular wall stress (HF severity). | – van der Meulen 2021: NT‐proBNP was the sole independent predictor in multivariate; a 2‐fold higher NT‐proBNP corresponded to about a 2.8‐fold higher risk of death/HTx.31 |
| Strong predictor of death/HTx: higher NT‐proBNP = higher risk; declining NT‐proBNP over time = improving hemodynamics/outlook. | – Persistent high NT‐proBNP trajectory in non‐survivors vs a decline in survivors over time.31 | |
| BNP (active hormone) | Similar to NT‐proBNP (acute HF load). Often used in acute settings; very high BNP = very high near‐term risk. | – Price 2006: children with 90‐day events had median BNP 735 pg/mL vs 37 pg/mL in others.38 |
| – BNP ≥300 pg/mL had HR about 63 for death/hospitalization/listing at 90 days.38 | ||
| Neutrophil–lymphocyte ratio (NLR) | Marker of systemic inflammation/stress response. Higher NLR = worse prognosis; reflects inflammatory activation and illness severity. | – Arici 2025: NLR significantly higher in children who died (median about 4.9) vs survivors (about 1.6); NLR >2.75 identified high risk (AUC about 0.79).12 |
| – NLR remained an independent mortality predictor in Cox analysis (HR about 2–3 per unit).12 | ||
| Systemic inflammation index (SII) | Captures inflammation + thrombocytosis: (neutrophils × platelets)/lymphocytes. | – Arici 2025: SII higher in non‐survivors; SII > 1.43 × 106 had AUC about 0.73 for mortality.12 |
| Higher SII = worse prognosis, similar interpretation to NLR. | – SII independently associated with mortality (p<0.05 in multivariate).12 | |
| Hyponatremia (low serum Na) | Indicates severe HF (neurohormonal activation → water retention). | – Arici 2025: deceased patients had lower Na (mean 132.5 vs 136.8 mmol/L, p=0.02); each 1 mmol rise in Na associated with about a 33% mortality risk reduction.12 |
| Low Na = worse prognosis; marker of advanced HF/poor perfusion. | – Consistent with adult HF: Na <134 is high risk; often a late HF sign. | |
| Troponin T/I | Reflects myocyte injury (acute damage). | – Arici 2025: troponin I did not differ significantly between survivors vs non‐survivors; troponin often normal/mildly elevated in chronic DCM.12 |
| Prognostic mainly in acute/fulminant settings (e.g., myocarditis; very high troponin implies higher acute risk/need for ECMO). In chronic DCM: not proven for long‐term risk stratification. |
Currently, among pediatric DCM cohorts, NT-proBNP is the biomarker with the most reliable prognostic power and it is often the main determinant even after the clinical and imaging covariates have been taken into consideration.31 Through a prospective multicenter repeated-measures design, the study has revealed that el-evated NT-proBNP levels were a very strong predictor of death/HTx and, most importantly, the longitudinal pat-tern contributed substantial prognostic information: the children that eventually reached the endpoints had the NT-proBNP levels gradually increasing (and staying high) compared to the patients without events, thus, encour-aging “dynamic risk” evaluation over merely relying on one baseline snapshot.31 Further support for the non-linearity of the relationship between NT-proBNP and risk, as well as the variation of this association with the time horizon and cohort characteristics, comes from registry-linked analyses which thus fortify the notion that the in-terpretation should be local and not connected to a sin-gle universal cutoff ( Table 3).37 BNP levels measured in pediatric outpatient clinics for chronic LV systolic dys-function patient groups (including cardiomyopathy phe-notypes) have shown excellent risk discrimination of chil-dren at high risk of adverse events in the near short-term ( Table 3).38 Essentially, the BNP/NT-proBNP are most effectively employed as (i) baseline severity markers and (ii) serial response-to-therapy markers, in which case a failure to decline or a rebound in elevations should raise a concern still higher and lead to a re-examination of the trajectory and escalation timing.31,37
2) Inflammatory Markers (Neutrophil-to-Lymphocyte Ratio (NLR) and Systemic In-flammatory Index (SII)):
CBC-derived indices could reflect systemic inflammatory activation and physiologic stress in pediatric DCM. In a re-cent study of the pediatric DCM cohort, the non-survivors had higher NLR and SII. Besides that, such indices show quite a good discrimination for mortality by the ROC anal-ysis, and more importantly, even after adjusting for other variables, these two parameters are still significantly cor-related with mortality within that dataset ( Table 3).12 The parameters are appealing as they both inexpensive and widely accessible, and thus, they could have poten-tial in environments that lack advanced biomarker assays. However, due to the fact that the current evidence base is still limited by the number of cohort and single-center de-sign, one should consider them as complementary signals that need to be validated multicenter.12
Markers of myocardial injury (troponins) and general in-flammation (e.g., CRP) have not been reliable prognostic tools in chronic pediatric DCM except for those cases with specific etiologies; in the same pediatric cohort, there was no significant difference between troponins and CRP in terms of outcome and they were not significant predictors in the univariate time-to-event analyses ( Table 3).12 Routine chemical imbalances in the body may still indi-cate more advanced conditions. For instance, a reduced level of sodium in the blood (serum sodium) was linked with death and demonstrated a signal for prognosis in the univariate analysis of a pediatric dilated cardiomyopathy (DCM) cohort, which is in line with hyponatremia being a sign of severe heart failure (HF) neurohormonal activa-tion and patient vulnerability ( Table 3).12 Care should be taken when interpreting observations of other routine measures (magnesium for example) as the sample size is small and there could be scale effects.12 Besides that, the severity of the biomarkers together with the physio-logical correlates can actually reveal quite a lot: In a tiny pediatric cohort, it was also found that non-survivors had lower transmitral E-wave velocity, thus, indirectly con-firming the idea that very low forward-flow surrogates might be typical of advanced dysfunction. Nevertheless, the discoveries have to be replicated in bigger datasets before they can be used clinically.12
D. Genetic Factors and Genotype-Phenotype Correlations
Genetic testing has fundamentally changed our understanding of the causes and outcomes of pediatric DCM. Today, an increasingly standard part of care is compre-hensive genomic testing, mainly because it can (i) iden-tify the cause of the disease, (ii) facilitate family mem-bers’ testing through cascade screening, and (iii) pro-vide risk assessment if the genotype, outcome associations are known in pediatric populations.11,39 Diagnostic yield varies among cohorts because of different pheno-type definitions (isolated vs. syndromic DCM), age com-position, and sequencing/interpretation pipelines. How-ever, various studies have demonstrated that a signifi-cant portion of children initially diagnosed as “idiopathic” cases, are eventually discovered to have a genetic basis for their condition when thoroughly examined.11,39 Studies of contemporary pediatric DCM cohorts show that the yield varies considerably between identifying genetic causes, and pediatric outcome data now reveal that the “gene-positive” status can have prognostic implications, i.e., it is not solely diagnostic. Genome sequencing of a nationwide Dutch pediatric DCM cohort (2010, 2017) un-covered LP/P variants in more than one-third of the chil-dren tested, and the carriers of the variants had a signif-icantly worse transplant-free survival, with a greater risk (hazard) of death/HTx during the follow-up period.11 One of the things that most clearly reveals the usefulness of clinical utility is a lot of sequencing work done in stud-ies of pediatric cardiomyopathy. For instance, the exome-based work in a large pediatric cohort found a molecular diagnosis in about a third of patients overall. The study also showed that the yield depended on the age and was lower in infants. This therefore supports the notion that the yield from a cohort is determined by its make-up and the risk interpretation given afterwards.40,41
i. Gene spectra and age-dependent patterns
One’s genetic background may be physiologically differ-ent at different ages and thus can be very useful for diag-nosis. The research group from the Netherlands found that the profile of the spectrum varies among the different age groups. At a very early stage, even since birth-onset cases of sarcomeric/cytoskeletal genes (e.g., MYH7 and others) dominated. Whereas, the children of older/adolescent groups generally had the involvement of more genes such as TTN and RBM20 in that pattern.11 Clinically, it also helps to calibrate the pre-test probability and also guides as to what to tell the patient: for in-stance, if the patient comes very early the doctor may start thinking of certain sarcomeric etiologies, but if the ven-tricular dysfunction develops during the teenage years, the genetic cause will be suspected to be one that leads to severe remodeling of the heart and increases the risk of arrhythmias as is generally known from the DCM ge-netics literature.11,39
ii. Presence of a pathogenic variant: prognosis is not purely “baseline DCM,” and genetics can modify “acquired” triggers
Identification of an LP/P variant offers a major cardiomy-opathic substrate which, at a population level, seems less capable of completely reversal than a truly tran-sient injury. The pediatric outcome data give direct backing to this: in the Dutch nationwide cohort, dur-ing follow-up, the variant carrier children had a signifi-cantly higher risk of death/HTx than the variant-negative children.11 In fact, the role of genetics also opens up new perspectives on the so-called “acquired” trig-gers, as it is a predisposition/second-hit model. A very good example that supports this notion comes from pe-diatric biopsy-proven myocarditis. In a cohort study, gene variants of cardiomyopathy were not uncommon; on the contrary, they were more frequent among those children who had a DCM phenotype, and thus, a signif-icantly worse event-free survival was found in children with gene variants. Therefore, part of the “myocarditis” presentation may be due to inherited susceptibility espe-cially when myocarditis is combined with DCM-like re-modeling.10 Convergent evidences drawn from adult population-based samples also demonstrate a higher fre-quency of rare cardiomyopathy-gene variants in acute my-ocarditis patients versus controls, hence genetically pre-disposed to be revealed by exposure to environmental or inflammatory stressors.42 It is always safe to be cau-tious about directly applying the generalizations made about adults to children, however, these signals give more weight to the argument that genomics should be evalu-ated early in cases of myocarditis complicated by the pres-ence of prominent DCM features or an atypical trajectory.10,42
iii. Specific genes and genotypes associated with poorer prognosis or “arrhythmic DCM” be-havior
While pediatric genotype, outcome datasets in DCM are inherently smaller than those of adults, modern pediatric cohorts and guidelines meta-analysis endorse the exis-tence of “arrhythmic” or higher-risk DCM genotypes (typ-ically including LMNA, FLNC, PLN, RBM20, and several others) that require an accelerated rhythm surveillance and individualized preventive strategy.11,39 Since pediatric quantification is limited in extent, it is most rea-sonable to accept that risk estimation is usually based on adult cohorts, whereas pediatric studies are now show-ing similar patterns, occasionally even early-onset severe disease.
iv. Genetic vs non-genetic DCM: recovery and risk-stratification implications
One practical combination which fits well with the current evidence is the idea that genotype status may explain to some extent why kids with the same baseline echocar-diography follow different paths. Disease-positive vari-ants may be less prone to continued reverse remodeling after the first injury, whereas negative-variant/idiopathic groups may even show a partial recovery in some cases, although there are exceptions and the cause of the dis-ease is still the main factor.43,11 Genotype can be seen as one of the factors influencing the decision of in-creasing the management intensity: it can serve to de-cide the necessity of more frequent follow-up visits, the implementation of more intensified rhythm monitoring, and referral to advanced heart failure programs at lower severity levels in high-risk/arrhythmic substrates cases, whereas, the choices of pediatric devices should be kept individualized and aligned with the guidelines.11,39 Finally, family screening becomes an urgent clinical out-come after the discovery of an LP/P variant. It allows for the identification of asymptomatic relatives and leads to better management of familial risk.11,39 Several pediatric DCM genotypes may produce phenotypes with a higher arrhythmic burden than others, and this vul-nerability is not fully captured by LVEF alone; therefore, genotype-informed rhythm surveillance appears more ap-propriate than relying on a single functional threshold. Table 4 picks out a few genes and the associated pheno-types/outcomes for easier reference:
| Genetic factor | Phenotype / outcome associations | Evidence |
|---|---|---|
| Any pathogenic variant (LP/P; gene‐positive) | Gene‐positive status is generally linked to a more severe phenotype and worse transplant‐free survival. Truly variant‐negative/idiopathic cases often do better mid‐term. | – Dutch cohort: LP/P carriers had about a 2.8‐fold higher risk of death/HTx vs variant‐negative (HR 2.8, 95% CI 1.3–5.8, p=0.007); events occurred at younger ages.11 |
| LMNA (Lamin A/C) | Early‐onset DCM with conduction disease and arrhythmias; high risk of malignant VT/VF/SCD and progression to end‐stage HF. | – High‐risk genotype: adult cohorts report SCD risk up to about 46% by age 30.46 |
| – Pediatric series show frequent pacer/ICD need and transplant in youth,47 with malignant arrhythmias in some children.48 | ||
| PLN (p.Arg14del) | Arrhythmogenic DCM/AC, often adolescent onset; high VT/VF risk even with mild dysfunction; progression to advanced HF is common. | – p.R14del linked to lethal ventricular arrhythmias; ICD often considered. Enriched in familial cases; treated as high‐risk in ESC guidance (ICD consideration, e.g., if LVEF <45%).49 |
| FLNC (truncating variants) | Arrhythmogenic cardiomyopathy overlapping DCM; can present in childhood with severe systolic dysfunction and arrhythmias; high SCD risk when phenotype expressed early. | – Baban 2022 (peds): early cardiac disease; about 21% had long‐QT/other arrhythmias; many required advanced care.50 Often managed as high‐risk (ICD once VT documented). |
| RBM20 | Typically severe, adolescent‐onset DCM with very low EF and high VT/VF risk; often rapidly progressive to end‐stage HF. | – Dutch data: RBM20 in about 6.7% of older pediatric DCM; aggressive course.11 Other reports show early onset, arrhythmias, and advanced HF support by young adulthood.51 Listed as high‐risk in ESC guidance.11 |
| TTN truncating variants (TTNtv) | Most common; variable penetrance/outcomes (some stabilize, others progress to transplant). Recovery is often limited if severe. | – Dutch: TTN most frequent (about 10%) in ages 2–18.11 Pediatric TTNtv linked to earlier diagnosis and often worse outcomes vs non‐TTN idiopathic, though a subset remains stable.43 |
| MYH7 | Common in infants/children; often early onset with dilated or hypertrabeculated LV; can be aggressive, especially with infant onset and low EF. | – de Frutos 2024 (n=44): 36% reached transplant/HF death; NYHA III–IV and LVEF ≤35% predicted progression.52 |
| Desmosomal genes (e.g., DSP) | Arrhythmogenic cardiomyopathy overlapping DCM; can mimic myocarditis (near‐normal EF early) then progress to dilation; arrhythmic risk can be substantial. | – Choi 2024: frequent VT/VF and myocarditis‐like episodes; many required antiarrhythmics/ICD; some progressed to DCM over time.53 |
Attempts to formalize multivariate risk prediction for pe-diatric DCM are currently underway, but are still not as developed as adult heart failure scores. The reason be-ing that pediatric cohorts are smaller and endpoints are more often considered as time-to-event outcomes (e.g., death or heart transplantation [HTx]).14 Clinicians in fact use essential predictors such as age at diagno-sis, severity of heart failure leading to hospital admis-sion, remodeling/ventricular dysfunction, and the etio-logical context to do a sort of informal multivariate risk formula even if there are no considered bedside scores.14,31 Recently, research has increasingly focused on cohort-derived point scoring tools. In a 2025 pedi-atric DCM study, Xu et al. created a nomogram and a streamlined score based on five predictors (age at onset, advanced functional class [III, IV], moderate-to-severe MR, low-voltage ECG, and requirement for vasoactive drugs) to forecast death/HTx, their results showed that the two methods had a discrimination of AUC 0.804 (nomogram) and AUC 0.794 (simplified score), respec-tively. A high-risk-cutoff ≥13.25 was proposed by the authors. At the same time, they stressed that external validation is a must before the model can be used rou-tinely.14 Methodologically, a significant step forward in line with the principle that the trajectory matters is the move to time-updated models that incorporate re-peated measurements rather than using baseline snap-shots only. Serial evolution of biomarkers and imaging features can improve separation between responders and non-responders, and dynamic markers may outperform a single baseline value for predicting adverse outcomes over follow-up.31 Similar methods focus on interme-diate trajectories that are closely connected to the down-stream risk, such as LV reverse remodeling after the ther-apy guided by the guidelines. As an illustration, Han et al. came up with a predictive model for reverse remod-eling in children with recent-onset DCM which can be used for more personalized counseling and determining the intensity of follow-up based on the expected remod-eling response.44 Lastly, machine-learning (ML) and data-driven phenotyping techniques are gaining ground in identifying subgroups with clinical significance that are not easily distinguishable by means of linear point scores.
Fu et al. characterized the phenotypes of patients with very different event risks in their electronic-record clus-tering study (n = 279) from 2025. The higher-risk phe-notype patients were at substantially elevated risk of ad-verse outcomes (reported HR 8.10, 95% CI 4.05, 16.19), “phenotype-driven” methods can be used to supplement standard regression, which, however, still has to be exten-sively externally validated and tested for transportability.45 Practical takeaway: the existing pediatric DCM risk look promising but the majority of them are lim-ited to single cohorts only and thus should be considered as decision-support prototypes until they get externally validated and calibrated across systems where’death vs HTx’ pathways differ.14,45 Representative prognostic modeling approaches in pediatric DCM, including score-based, time-updated, reverse-remodeling, and machine-learning approaches, are summarized in Table 5.
| Model / approach | Cohort (design) | Target outcome | Key inputs | Reported performance |
|---|---|---|---|---|
| Nomogram + simplified bedside score14 | Single‐center pediatric DCM (n=233; 2019–2024)14 | Death or heart transplantation14 | Age at onset; Ross/NYHA III–IV; moderate–severe MR; low‐voltage ECG; vasoactive drug requirement14 | AUC about 0.804 (nomogram) and 0.794 (simplified score); cutoff ≥13.25 for higher risk.14 |
| Time‐updated repeated‐measures model31 | Prospective multicenter pediatric DCM (n=137) with serial sampling31 | Death or heart transplantation31 | Serial NT‐proBNP trajectories (time‐updated)31 | Doubling NT‐proBNP → HR 2.78 for death/HTx after adjustment.31 |
| Reverse‐remodeling prediction model44 | Recent‐onset pediatric DCM (n=146) treated pharmacologically44 | LV reverse remodeling after therapy44 | Age at diagnosis; LVEDD z‐score; QRS duration44 | C‐index 0.903; AUC 0.907 in development cohort.44 |
| ML phenomapping (clustering) + classifier45 | Pediatric DCM EHR cohort (n=279)45 | Adverse events over follow‐up (death/HTx composite)45 | EHR‐derived multi‐domain variables; phenotype membership used for classification45 | High‐risk phenotype HR about 8.10; classifier AUC about 0.86.45 |
In this narrative review, we brought together a wide and clinically pertinent range of prognostic factors for pediatric dilated cardiomyopathy (DCM). These fac-tors cover the baseline patient context, the severity of the presentation, cardiac imaging, biomarkers, and ge-netic/arrhythmic modifiers. Throughout, the consistent main message is that pediatric dilated cardiomyopathy (DCM) should not be considered a single disease entity. It is a heterogeneous syndrome with multiple biological and clinical trajectories. Thus, valuable prognostication is hardly ever based on just one measurement; it is more the case that the initial risk factors, the objectively mea-sured severity, and, most importantly, the first phase of the disease progression has to be combined.
Among current cohorts and the latest imaging meta-analyses, three features derived from imaging repeatedly serve as the basis for risk: left ventricular (LV) systolic dysfunction, LV dilation (LVEDD z-score), and severe func-tional mitral regurgitation (MR). These markers indicate an increased disease burden and adverse remodeling, and they have been repeatedly associated with death, ventric-ular assist device (VAD) implantation, or heart transplan-tation (HTx). Significantly, their prognostic interpretation is based on the amount and time: deep initial damage is an indicator of high risk at early time, while the way the structure changes during follow-up usually identifies risk more precisely than initial severity. Patient profiles and the cause of disease determine “baseline biology”, which in turn significantly impacts the likelihood of recovery or progression. The older the age at the time of diagnosis, es-pecially if the disease is idiopathic/familial or no obvious reversible trigger is found, the more it is associated with a poorer prognosis in terms of transplant-free survival com-pared to infancy. In contrast, infants, initially presenting with myocarditis or tachyarrhythmia-related dysfunction, may demonstrate a greater capacity for recovery on con-dition that they survive the initial high-risk phase. But at the same time, modern data also warn against too sim-plistic a divide: phenotypes presenting with myocarditis are not always a good prognosis, and cardiomyopathy-associated pathogenic variants can be found in myocardi-tis and be linked to unfolding similar to genetic DCM. The overlap here reinforces the idea that the etiology assign-ment should be reconsidered if the clinical course is un-usual and it also supports the idea of early referral to ge-netic evaluation in certain cases. One of the major factors that influence the prognosis of a patient is the severity of clinical heart failure (HF) when they first present. Chil-dren who have symptoms of very severe heart failure (CF Ross/NYHA III, IV), are in shock, or need ICU/inotropes are in great danger in the short term and, thus, their treatment is usually initiation of early-stage treatment rather than further outpatient optimization of these chil-dren. On the other hand, mild symptoms (class I-II), par-ticularly in potentially reversible etiologies, are less fre-quently linked to an early death; however, the long-term outcomes still rely on remodeling and biomarker trajecto-ries. These findings correspond to the fact that children’s risk is mostly “front-loaded” immediately after diagnosis and, later on, it is more and more influenced by chronic remodeling and need for arrhythmic vigilance.
In order to transform diverse prognostic data into practi-cal clinical insights, we have suggested a comprehensive multi-layered framework for the risk stratification of pedi-atric DCM patients ( Figure 1). At the time of a diagnosis, it is necessary to assess the risk through profiling in four domains that interact with each other: baseline context (age, etiology, genotype/family history and resource set-ting), severity of the presentation, phenotype/remodeling derived from imaging, and biomarkers. Risk is further de-fined through a formal review of the changed trajectory in the early follow-up period (usually 1, 3 months and 6, 12 months), focusing on the changes in LV size/function (z-scores), MR, serial natriuretic peptides, clinical course, and growth. Electrical and genetic factors not only de-termine the course of the disease but also the final re-sult, especially in terms of the risk of arrhythmias. From such a strategy, two main clinical pathways can be identi-fied: a high-risk path (extensive/progressive dysfunction and no improvement in the biomarker pattern) that leads to death/VAD/HTx and a small percentage of arrhyth-mic events, and a recovery path (remodeling reversal and biomarker lowering) that indicates good outcomes but at the same time is conscious of relapse risk even after ap-parent normalization. An advantage of this model is that it allows very easy decision-making, it mirrors the most trustworthy evidence signals (imaging + NT-proBNP + se-rial change), and and it reflects the real-world variation in access to diagnostic resources.
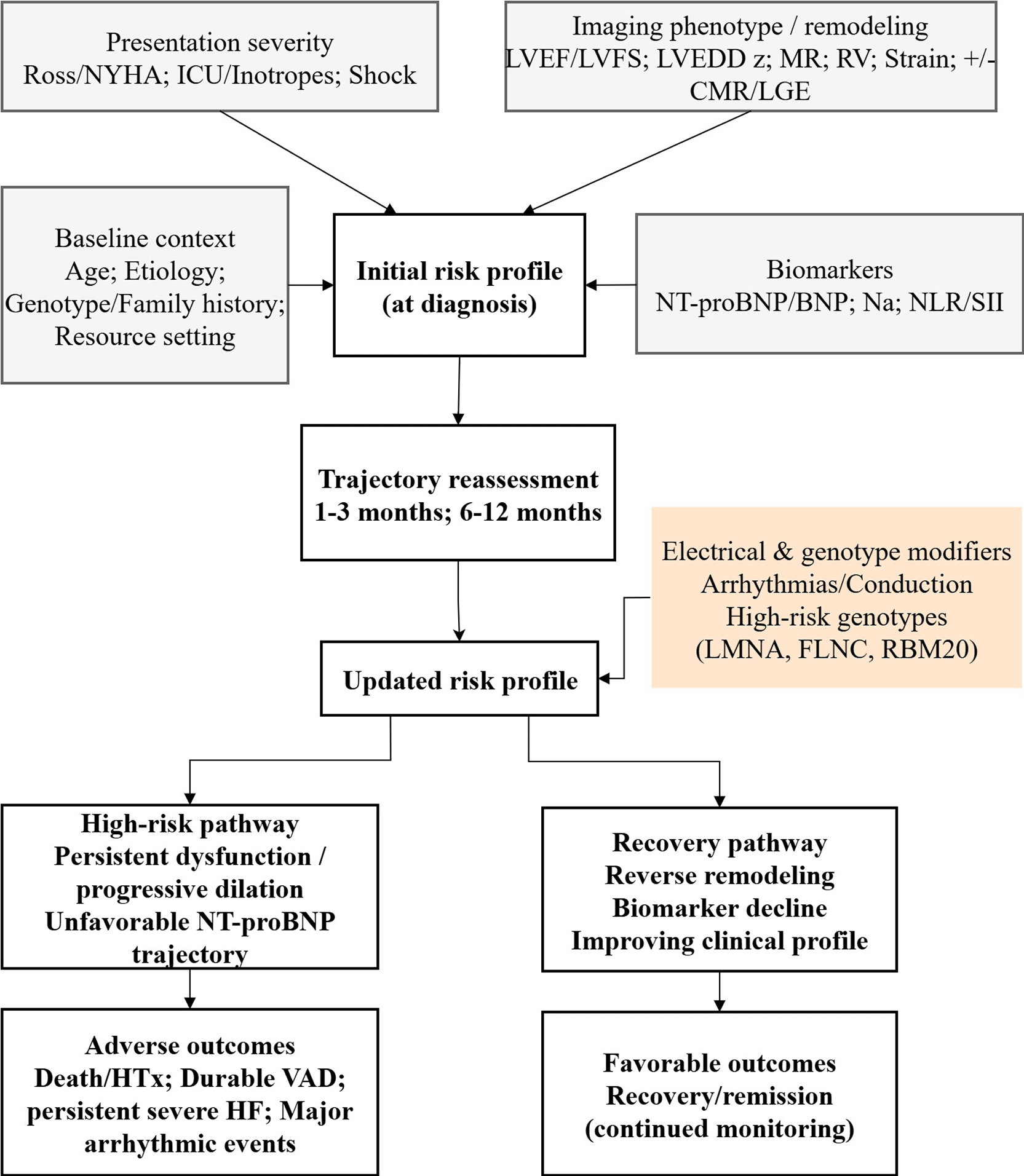
Among circulating biomarkers, NT-proBNP is always the one that most accurately reflects the hemodynamic stress and neurohormonal activation. Besides, it is often found to be a powerful independent predictor of death/HTx. Serial interpretation is where its greatest value lies, as trajectories (decline versus persistently high levels) give an objective indication of response to therapy and chang-ing risk. Alongside that, we also pointed out inflamma-tory indices like NLR and SII that have been identified as emerging prognostic adjuncts in the latest pediatric co-horts. Currently, these markers ought to be seen as sup-portive considering that they can be influenced by an in-tercurrent infection and physiological stress and shouldn’t be utilized as independent decision thresholds.
Combining genotype and family history is becoming more and more significant for the prognostic reasoning of pedi-atric DCM. A gene-positive status has been less favourable in terms of prognosis in various national cohorts as com-pared to gene-negative cases, and gene-specific patterns may alter the predominant way of risk. In the clinic, it is prudent to be cautious, but one can still proceed with a few steps: Genetic results will certainly be of help in refining the intervals of monitoring and screening of rela-tives, and they may justify an earlier surgery in cases when the phenotype is extremely severe, but the main criteria of the decisions should be the severity of the child’s phe-notype and its development rather than solely the geno-type. Arrhythmic outcomes account for a smaller propor-tion of events than progressive HF/HTx; however, they remain clinically important because, in selected high-risk children, they may be preventable. Currently, there is no pediatric evidence that supports simple extrapolation of adult primary-prevention ICD thresholds based solely on LVEF; rather, the best way to evaluate arrhythmic risk is by combining the electrical history (sustained ventricular arrhythmia, syncope concerning for arrhythmia, conduc-tion disease) structural severity, trajectory, and genotype if available.
This synthesis of prognosis points has direct care implica-tions:
1. Timing of referral and escalation: Children coming with advanced HF at presentation, severely impaired LV function/dilation, severely MR, still very high or non-declining NT-proBNP, or a bad early course should lead to early referral to advanced HF/HTx centers and initiation of escalation pathways (includ-ing consideration of VAD/HTx evaluation where ap-propriate).
2. Device decisions: ICD in pediatric DCM should be considered individually and not based on a single EF cut-off only. Other factors for consideration are ar-rhythmic history, phenotype severity, trajectory, and genotype-informed risk where relevant.
3. Monitoring intensity along with supportive care: the disease outcome or prognosis should guide the reg-ularity of the follow-up visits, the need for prompt therapeutic changes, and the various support areas such as nutritional intervention in children experi-encing growth failure. Repeated echocardiography along with unchanged z-score measurement and re-peated natriuretic peptides are only a few practical ways of continuous risk assessment.
4. Family counselling: Objective, quantifiable measures (imaging severity, biomarkers, trajectory) not only enable straightforward yet nuanced counselling but also serve as a rationale for the escalation of ther-apy; whereas reverse remodeling and biomarker re-duction may offer cautious hope, provided that reg-ular follow-up is maintained.
5. Context-of-care: prognostic interpretation should quite clearly take into account access to advanced di-agnostics and therapies; risk model tools which are based on transplant-capable environments may not totally transfer to resource-limited settings.
Most of the pediatric DCM evidence base is still largely composed of observational studies and also cohort het-erogeneity along with changing treatment eras. A lot of predictors are interrelated (collinearity), so it is not very clear how to separate the independent effects of each and the risk of “double counting” closely related mea-sures is also increased. There are several new markers (e.g., inflammatory indices and advanced CMR fibrosis measures) which are still based only on relatively small pediatric samples and thus require multicenter valida-tion. Moreover, the modelling challenge is made even more complicated due to the fact that both transplanta-tion and VAD, by resulting in patients bypassing their nat-ural progression, yield the observation of event rates that reflect referral patterns and resource availability. To sum up, although psychosocial and system factors (such as ac-cess barriers, adherence, and follow-up constraints) are scarcely recorded, they can significantly change the real-world risk.
Priorities set forth for the field are: (i) to carry out ex-ternal validation and transportability testing of already available pediatric risk tools in different settings; (ii) stan-dardizing definitions and handling of endpoints, includ-ing competing risks; (iii) prospectively harmonizing imag-ing and biomarker protocols in multicenter studies (this also includes strain and natriuretic peptide sampling); (iv) genotype-informed cohorts powered for gene-specific pediatric risk estimation and myocarditis, genotype inter-actions; and (v) implementation studies to assess whether structured, trajectory-based risk stratification improves referral timing and family-centered outcomes.
In summary, the prediction of outcomes in pediatric DCM has evolved from the reliance on single markers to an inte-grated, trajectory-based view that includes genotype, car-diac phenotype and remodeling, circulating biomarkers, and system-level modifiers such as nutrition and resource context. In this review, we have put together these lay-ers into a unified framework that is helpful for making a working-level risk stratification at the time of diagno-sis and its refinement during the early period of follow-up, thus assisting clinicians in telling the difference be-tween children who need quick escalation of their treat-ment and those who may be kept under observation with cautious optimism. Most importantly, prognostic factors should not be considered as fixed destinies; they only act as decision-making aids which ought to be accompanied by prompt actions. With a high-risk profile, targeted in-tervention should be intensified, the patient should be more closely monitored, and referral to advanced heart failure/transplant pathways should be done early if ap-propriate, all with the main aim of changing the child’s path. Advances in imaging, biomarkers, and genomics, along with external validation and putting into practice risk-guided pathways, promise to improve survival and quality of life for children with dilated cardiomyopathy.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)