1Department of Biostatistics, Harvard School of Public Health, Boston MA, 02215, USA 2Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston MA, 02215, USA 3Ludwig Center for Cancer Research at Dana-Farber Cancer Institute and Harvard Medical School, Boston MA, 02215, USA 4Center for Sarcoma and Bone Oncology, Dana-Farber Cancer Institute, Boston MA, 02215, USA 5Current affiliation: Division of Biostatistics & Bioinformatics, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, 21205-2013 , USA 6Current affiliation: Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, 21205-2179 , USA
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Background: Most patients with advanced gastrointestinal stromal tumors (GIST) develop drug resistance to tyrosine kinase inhibitors (TKIs) within two years of starting therapy, whereas most chronic myeloid leukemia (CML) patients in chronic phase still exhibit disease control after a decade on therapy. This article aims to explain this divergence in long term outcomes. Methods and results: By combining clinical and experimental observations with mathematical formulas we estimate that, in advanced GIST, the genetic changes responsible for resistance are generally already present at disease detection. Conclusion: This result has relevant clinical implications by providing support for the exploration of combination therapies.
Corresponding author:
Cristian Tomasetti
Competing interests:
CT and GP have no competing interests to disclose. GDD discloses the following: Novartis. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Pfizer. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Infinity Pharmaceuticals. Consultant, <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Regulatory presentation support, uncompensated. Glaxo Smith Kline. Consultant, <$10k per annum. Kolltan Pharmaceuticals. Chair, Scientific Advisory Board. Chair, Medical Advisory Board. Consultant. Equity (minor stake, non-public).
Grant information:
The work of CT was supported in part by the National Institute of Health under Grant T32 CA009337. GDD was partially funded by DFHCC SPORE (NCI Grant) for GIST research, as well as Ludwig Center for Cancer Research at Dana-Farber/Harvard Cancer Center. GP was supported in part by the NIH/NCI grant 5P30 CA006516-46.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Gastrointestinal stromal tumors (GIST) are sarcomas arising in the muscle wall of the gastrointestinal tract. The majority of GISTs are driven by activating mutations in the receptor tyrosine kinases KIT or PDGFR-α, whose aberrant signaling induces uncontrolled proliferation and decreased apoptosis1. Because of the small number of driving genetic mutations, GISTs represent a paradigm for kinase-driven solid tumors, and offer one of the best models to shed light on fundamental questions about cancer, providing a critical understanding of more genomically complex solid tumors.
Tyrosine kinase inhibitors (TKIs), such as imatinib and sunitinib, block the aberrant activation of KIT, leading to major clinical benefits of objective response and durable disease control. Imatinib represents the standard first-line therapy for this disease when it is surgically incurable. However, imatinib does not cure advanced GIST2. Moreover, while the large majority of patients treated with imatinib do not show signs of primary resistance (disease progression within the first six months of therapy), secondary resistance to imatinib emerges in at least half the patients after two years of therapy and in more than 80% of patients after seven years1,3,4.
Imatinib was originally introduced to treat chronic myeloid leukemia (CML), because of its specificity for the TK domain in the bcr-abl translocation gene, which characterizes the disease. Imatinib represents the standard first-line therapy and has led to dramatic improvements in outcomes for CML patients in the chronic phase: at six years the estimated event-free survival is 83%, and an estimated 93% of treated patients are free from progression5. Importantly, secondary resistance is far less frequent in CML5.
In this article we consider two fundamental, clinically relevant, but still unanswered questions: Why do patients with advanced GIST often relapse and does secondary resistance originate before or during TKI treatment?
Materials and methods
In order to obtain estimates for the probability of at least one drug resistant GIST cell being present at the time the tumor reaches a given diameter, and before the introduction of the TKI, we used formula (5) in Tomasetti et al.6:
This formula estimates the probability of having resistant mutants PR in a tumor of size M, (number of cells), and is derived by counting the number of divisions required for the tumor to reach that size. It is assumed that, at each cell division, there is a small probability u that one of the daughter cells is hit by a mutation known to induce drug resistance. The parameter l and d are the birth and death rates for the cell population, while a and b are the probabilities for the cells’ mode’s of division. This mathematical result is based on standard assumptions and has been successfully used to predict the development of acquired resistance to targeted epidermal growth factor receptor (EGFR) blockade in colorectal cancer7. In order to apply this formula we used parameter estimates available in the literature, as follows.
The somatic point mutation rate u can be estimated to be between 10-9 and 10-8 per base per cell division8,9. There are many known point mutations causing resistance to imatinib10. To obtain a conservative bound, we assumed a value of 10 point mutations in our calculations, though the actual number could be higher11. Overall, the probability of a point mutation causing drug resistance in GIST should then be at least 10-8 per cell division. It has been estimated that 10-9 cancer cells are present per cm3 of tumoral mass12. As our goal was to generate an unfavorable scenario to our hypothesis, and since stromal tissue and other types of cells may be present, we halved this amount. Long-term drug resistance requires secondary mutations to be present in cells that are long-lived or able to self-renew13. KIT is known to have anti-apoptotic activity, thus contributing to cell lifespan14. Alternatively, resistance could arise from mutations in rare cells possessing stem cell attributes. Interstitial cells of Cajal progenitors are a potential candidate15. Their frequency is estimated to be 6.2 × 10-3 of all cells16. To be conservative, we only considered the stem cell compartment and set all other parameters of the main formula (5) in Tomasetti et al.6 to zero.
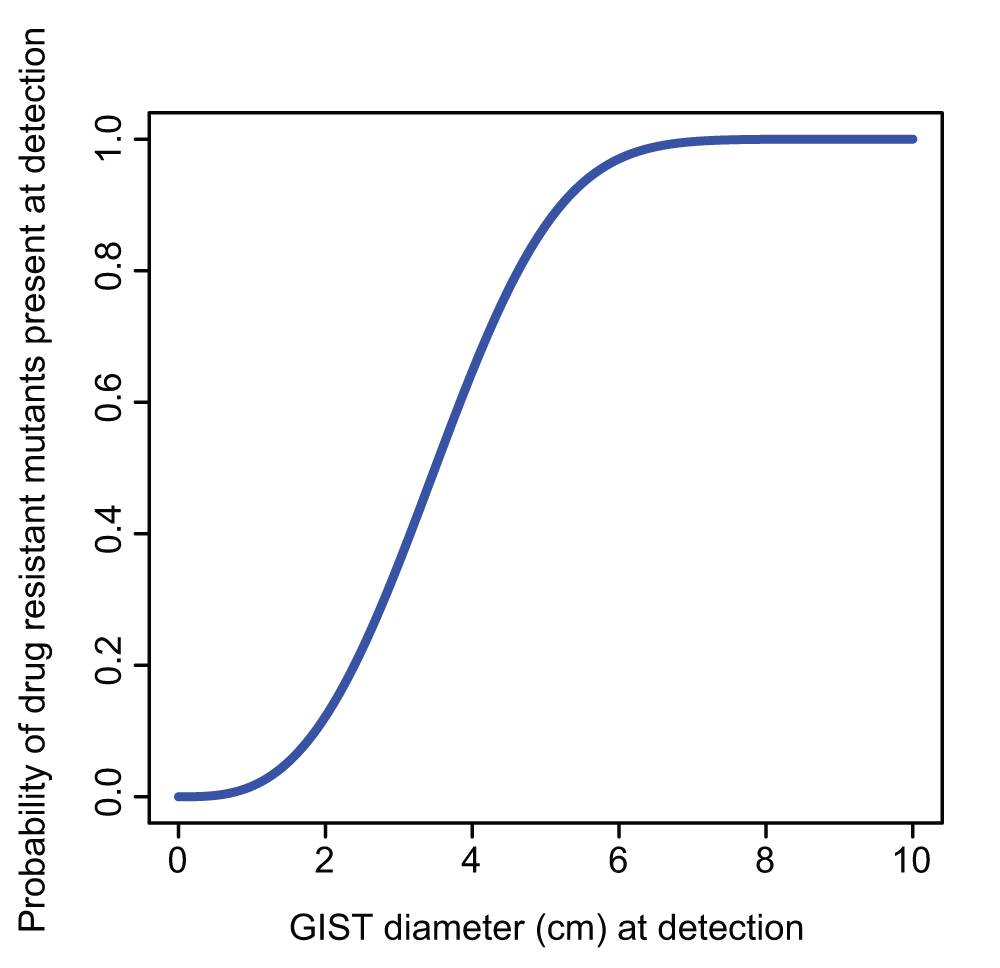
Using these results and parameters values, we can derive the lower bound of Figure 1 for the probability of at least one drug resistant GIST cell being present at the time the tumor reaches a given diameter, and before the introduction of the TKI. Because we selected parameters to generate an unfavorable scenario to our hypothesis, we expected the actual curve to be above the curve of Figure 1. The calculation and the figure were obtained using the freely available R software (version 2.15.3)17.
Figure 1. The estimated lower bound for the probability that mutant cells resistant to imatinib are already present in a gastrointestinal stromal tumor (GIST) at time of treatment, as a function of tumor diameter.
Results
We combined experimental data with our mathematical formulas6 that recently have been successfully used to predict the development of acquired resistance to targeted EGFR blockade in colorectal cancer7. By so doing, we have obtained the relationship shown in Figure 1 between the tumor diameter at detection, and the probability that the tumor already harbors a resistant mutant at that time (see Materials and methods for the derivation of Figure 1). For example, for GISTs with diameters of 2 and 6 cm we estimate this probability to be equal to 0.12 and 0.97, respectively.
We now bring these elements to bear in interpreting recent clinical observations. The size of a GIST at presentation may vary between 1 and >40 cm in diameter1. For example, in a clinical trial (NCT00237185) of 147 patients with unresectable or metastatic GIST expressing KIT and treated with imatinib, 75% of the patients had a GIST whose diameter was larger than 7 cm at treatment18. Based on Figure 1, we would expect the large majority of these patients to have resistant mutant cells already present by the time of GIST detection and therefore to develop resistance during therapy. In fact, only about 20% of the patients in this clinical trial were still progression-free after five and a half years from the start of imatinib treatment18.
The combination of experimental and clinical data with our mathematical estimates implies then that secondary resistance in patients with advanced GIST is to be expected and is due to the large size of the tumor at detection time. That is, mathematical estimates indicate that mutations responsible for secondary resistance are already present before the start of the treatment in patients with large tumor sizes, a key factor in explaining the high relapse of advanced GIST patients to TKI.
Discussion
It has been recently suggested that secondary resistance may be the result of the treatment itself rather than pretreatment mutations4. It is worth considering this issue further. Secondary mutations are not usually found at detection using conventional Sanger sequencing techniques4. However, these techniques are not sensitive for rare events. Because imatinib has not yet selected for resistant mutants, they may remain extremely rare, and thus are likely missed by the assays used for mutation analysis. Thus, the fact that mutants are rarely found at detection does not contradict the possibility that the point mutations did occur before treatment. Using our mathematical formulas again4, we estimate that if resistance is present in a GIST of 2 cm in diameter, only approximately 1 out of 108 tumor cells will be drug resistant. Most importantly, while resistance may also originate during treatment, this does not exclude the fact that random point mutations can occur during the pre-treatment phase at each cell division. We have shown that the estimated number of resistant mutants produced before the start of the treatment is sufficient to explain the observed clinical data. This logic also holds for the development of tertiary resistant clones that emerge within, on average, six months of starting second-line therapy with sunitinib following failure of imatinib19. Thus, our calculations, while not addressing the problem of primary resistance, are able to explain secondary resistance in advanced GIST.
Our prediction that patients with smaller GISTs at detection will have a smaller probability of progression has been already supported by the clinical correlation of tumor size at presentation with improved outcomes on imatinib18. With sufficient data, it may also be possible to observe a relation between tumor diameter at detection and probability of resistance that is similar in shape to Figure 1, since a resistant clone originating before therapy should eventually grow to a detectable size. Lastly, in the future, with next-generation DNA sequencing techniques, it may become possible to detect the presence of secondary mutations that represent a very small fraction of cells at initial presentation.
Our results, if confirmed, would have important clinical implications. A therapy using a combination of TKIs (for example by adding a new agent with novel spectrum activity to imatinib) would select only for those cells which have been hit by two point mutations, each causing resistance to one of the drugs. We estimate that such a probability is very small for realistic values of the GIST diameter (not considering cross resistance). Therefore, combination therapy could drastically reduce the development of drug resistance caused by point mutations. Also, earlier detection of GIST offers the potential to provide measurable improvements in outcome, as long as it can result in a sufficiently large reduction in the average diameter at detection.
Author contributions
CT and GP conceived the idea. CT, GDD, and GP designed and performed research, and wrote the manuscript. CT contributed the mathematical analysis.
Competing interests
CT and GP have no competing interests to disclose. GDD discloses the following: Novartis. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Pfizer. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Infinity Pharmaceuticals. Consultant, <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Regulatory presentation support, uncompensated. Glaxo Smith Kline. Consultant, <$10k per annum. Kolltan Pharmaceuticals. Chair, Scientific Advisory Board. Chair, Medical Advisory Board. Consultant. Equity (minor stake, non-public).
Grant information
The work of CT was supported in part by the National Institute of Health under Grant T32 CA009337. GDD was partially funded by DFHCC SPORE (NCI Grant) for GIST research, as well as Ludwig Center for Cancer Research at Dana-Farber/Harvard Cancer Center. GP was supported in part by the NIH/NCI grant 5P30 CA006516-46.
Faculty Opinions recommended
References
1.
Corless CL, Heinrich MC:
Molecular pathobiology of gastrointestinal stromal sarcomas.
Annu Rev Pathol.
2008; 3: 557–86. PubMed Abstract
| Publisher Full Text
2.
Le Cesne A, Ray-Coquard I, Bui BN, et al.:
Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial.
Lancet Oncol.
2010; 11(10): 942–9. PubMed Abstract
| Publisher Full Text
3.
Antonescu CR, Besmer P, Guo T, et al.:
Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation.
Clin Cancer Res.
2005; 11(11): 4182–90. PubMed Abstract
| Publisher Full Text
4.
Antonescu CR:
The GIST paradigm: lessons for other kinase-driven cancers.
J Pathol.
2011; 223(2): 251–61. PubMed Abstract
| Publisher Full Text
5.
Hochhaus A, O'Brien SG, Guilhot F, et al.:
Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia.
Leukemia.
2009; 23(6): 1054–61. PubMed Abstract
| Publisher Full Text
6.
Tomasetti C, Levy D:
Role of symmetric and asymmetric division of stem cells in developing drug resistance.
Proc Natl Acad Sci U S A.
2010; 107(39): 16766–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
7.
Diaz LA Jr, Williams RT, Wu J, et al.:
The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers.
Nature.
2012; 486(7404): 537–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
8.
Araten DJ, Golde DW, Zhang RH, et al.:
A quantitative measurement of the human somatic mutation rate.
Cancer Res.
2005; 65(18): 8111–7. PubMed Abstract
| Publisher Full Text
9.
Drake JW, Charlesworth B, Charlesworth D, et al.:
Rates of spontaneous mutation.
Genetics.
1998; 148(4): 1667–86. PubMed Abstract
| Free Full Text
10.
Heinrich MC, Corless CL, Blanke CD, et al.:
Molecular correlates of imatinib resistance in gastrointestinal stromal tumors.
J Clin Oncol.
2006; 24(29): 4764–74. PubMed Abstract
| Publisher Full Text
11.
Wang WL, Conley A, Reynoso D, et al.:
Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor.
Cancer Chemother Pharmacol.
2011; 67(Suppl 1): S15–24. PubMed Abstract
| Publisher Full Text
12.
Klein M, Bartoszynski R:
Estimation of growth and metastatic rates of primary breast cancer. In Mathematical Population Dynamics. (eds. Arino, O., Axelrod, D.E. & Kimmel, M.) (Marcel Dekker, New York, 1991).
13.
Tomasetti C:
A new hypothesis: imatinib affects leukemic stem cells in the same way it affects all other leukemic cells.
Blood Cancer J.
2011; 1(5): e19. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
Rossi F, Ehlers I, Agosti V, et al.:
Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor.
Proc Natl Acad Sci U S A.
2006; 103(34): 12843–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Bardsley MR, Horváth VJ, Asuzu DT, et al.:
Kitlow stem cells cause resistance to Kit/platelet-derived growth factor alpha inhibitors in murine gastrointestinal stromal tumors.
Gastroenterology.
2010; 139(3): 942–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Lorincz A, Redelman D, Horváth VJ, et al.:
Progenitors of interstitial cells of cajal in the postnatal murine stomach.
Gastroenterology.
2008; 134(4): 1083–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
R_Core_Team. R: A Language and Environment for Statistical Computing. 2.15.3 edn (R Foundation for Statistical Computing, Vienna, Austria, 2013). Reference Source
18.
Blanke CD, Demetri GD, von Mehren M, et al.:
Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT.
J Clin Oncol.
2008; 26(4): 620–5. PubMed Abstract
| Publisher Full Text
19.
Demetri GD, van Oosterom AT, Garrett CR, et al.:
Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial.
Lancet.
2006; 368(9544): 1329–38. PubMed Abstract
| Publisher Full Text
1
Department of Biostatistics, Harvard School of Public Health, Boston MA, 02215, USA 2
Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston MA, 02215, USA 3
Ludwig Center for Cancer Research at Dana-Farber Cancer Institute and Harvard Medical School, Boston MA, 02215, USA 4
Center for Sarcoma and Bone Oncology, Dana-Farber Cancer Institute, Boston MA, 02215, USA 5
Current affiliation: Division of Biostatistics & Bioinformatics, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, 21205-2013 , USA 6
Current affiliation: Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, 21205-2179 , USA
CT and GP have no competing interests to disclose. GDD discloses the following: Novartis. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Pfizer. Consultant, <$10k per annum. Honorarium <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Infinity Pharmaceuticals. Consultant, <$10k per annum. Research support to Dana-Farber for specific clinical trial agreements in our sarcoma unit, >$200,000 per year. Regulatory presentation support, uncompensated. Glaxo Smith Kline. Consultant, <$10k per annum. Kolltan Pharmaceuticals. Chair, Scientific Advisory Board. Chair, Medical Advisory Board. Consultant. Equity (minor stake, non-public).
The work of CT was supported in part by the National Institute of Health under Grant T32 CA009337. GDD was partially funded by DFHCC SPORE (NCI Grant) for GIST research, as well as Ludwig Center for Cancer Research at Dana-Farber/Harvard Cancer Center. GP was supported in part by the NIH/NCI grant 5P30 CA006516-46.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Tomasetti C, Demetri GD and Parmigiani G. Why tyrosine kinase inhibitor resistance is common in advanced gastrointestinal stromal tumors [version 1; peer review: 2 approved]. F1000Research 2013, 2:152 (https://doi.org/10.12688/f1000research.2-152.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Tomasetti et al. were combining clinical and experimental observations with mathematical formulas to estimate that in advanced GIST, the genetic changes responsible for resistance are already present from the beginning of treatment. The model is of highest interest, especially in
... Continue reading
Tomasetti et al. were combining clinical and experimental observations with mathematical formulas to estimate that in advanced GIST, the genetic changes responsible for resistance are already present from the beginning of treatment. The model is of highest interest, especially in respect of second and third line treatment with targeted therapies. However, prospective validation is required before this model can be incorporated in combination drug trials.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
This is a brief report depicting a mathematical model based estimation of presence of chemoresistant cells in gastrointestinal stromal tumor (GIST). The model incorporates the notion of the presence of rare cells possessing stem cell attributes (cancer stem cells) among
... Continue reading
This is a brief report depicting a mathematical model based estimation of presence of chemoresistant cells in gastrointestinal stromal tumor (GIST). The model incorporates the notion of the presence of rare cells possessing stem cell attributes (cancer stem cells) among heterogeneous cells within such mass, contributing to the chemotherapeutic resistance. On the basis of genetic point mutation as a source for the resistance, the probability formula for existence of resistant cells at a tumor size was constructed. Despite some constraints, the article presents reasonable assumptions providing for the lower bound of the probability. Based on this model, many of the clinical index GIST mass has significant likelihood of containing a tyrosine kinase inhibitor resistant cell at the time of the discovery. If future clinical findings support the assumptions made, the model would demonstrate a rational for combination therapeutics.
Specific points:
Title and Abstract: Appropriate
Article content: Methods and approaches are described appropriately and references made to the main formula utilized
Conclusions: Appropriate
Data: Appropriate as included
Minor item:
Materials and Methods second paragraph, 5th sentence should have “109 cancer cells” instead of “10-9 cancer cells are present per cm3 of tumor mass”.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)