Keywords
Recombinase mediated cassette exchange; RMCE; ROSA26; ES cells; Knock in; transgenic mouse; gene targeting; GFP
Recombinase mediated cassette exchange; RMCE; ROSA26; ES cells; Knock in; transgenic mouse; gene targeting; GFP
Transgenic mice created by pronuclear injection of a transgene have been a valuable resource for studying gene effects in a whole animal context, albeit there are several pitfalls of this technique. A well-known drawback was the randomness of transgene insertion where an endogenous gene or important regulatory element might be disrupted, resulting in a phenotype independent of the transgene of interest. Usually, multiple founders had to be tested and characterized before a clean transgenic mouse line could be established. To circumvent the inherent defect of this type of passive transgenesis, several approaches have been developed1 that allow transgenes to be inserted at defined genomic loci, such as ROSA26. These active forms of transgenesis can be achieved by utilizing a site-specific recombination system2 to facilitate the knock-in of the gene of interest. We have developed a toolkit (a ROSA26-tagged mouse embryonic stem (ES) cell line and two exchange vectors) for efficient gene knock-in at the ROSA26 locus in mouse ES cells by recombinase-mediated cassette exchange (RMCE). Standard microinjection of the knock-in ES cells would subsequently produce a chimera and the knock-in mice expressing the gene of interest, constitutively or conditionally, depending on which exchange vector is used.
First, we generated a mouse ES cell line, designated as R26FNF3-1F1 (1F1), in which the ROSA26 allele was targeted with a FRT and F3 flanked phosphoglycerate kinase (PGK) promoter and neomycin (Neo) selection cassette by homologous recombination in EC7.1 ES cells (Figure 1A), a hybrid strain with a background of C57BL/6 and 129X1/SvJ3. A targeting construct was made by the subcloning of a 5.8 kb BamHI fragment from the BAC clone RP23-401D9; a FRT-PGK-neo-F3 cassette amplified from PL451 (a gift from NCI Frederick) was inserted into the XbaI site so that the left and right homologous arms were both 2.9 kb in length. The PGK-neo marker was cloned in an opposite direction with respect to the ROSA26 locus. EC7.1 ES cells (5 x 106)3 were electroporated with 20 µg of linearized targeting construct and a total of 192 clones were screened for the homologous recombination event by long-range PCR. The PCR products were digested with specific enzymes (EcoRV for the left arm and ScaI for the right arm) to confirm the identities. A total of 7 targeted clones were recovered; clone R26FNF3-1F1 was established and was used in the subsequent experiments.
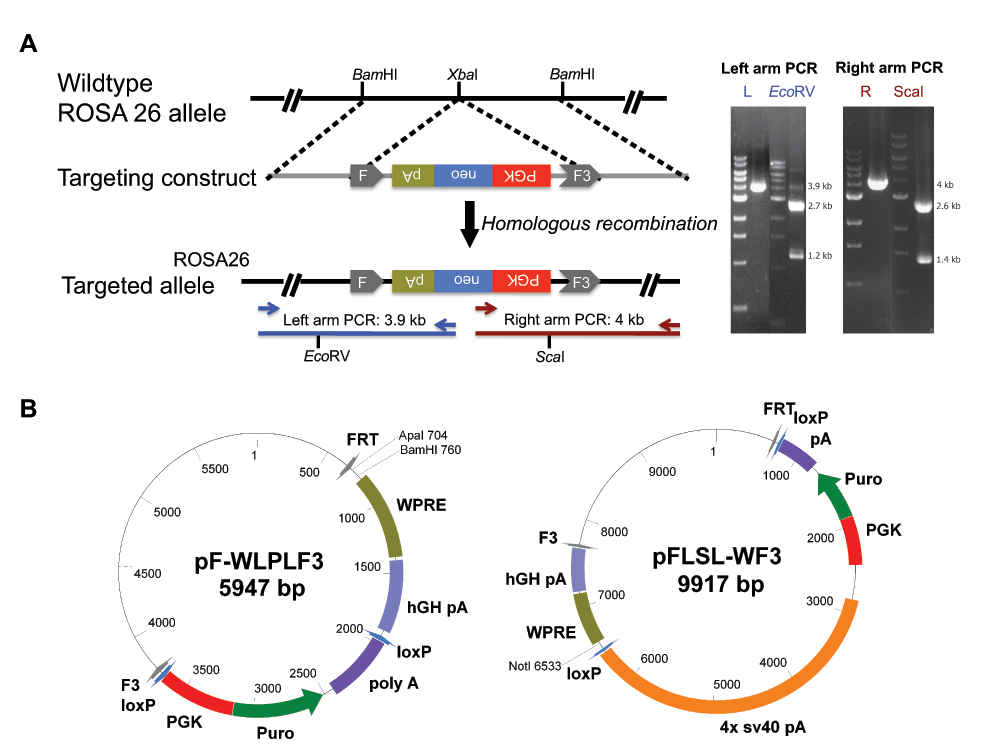
(A) Targeting the ROSA26 allele. The ROSA26 allele was targeted with a FRT and F3 flanked PGK promoter and neomycin selection cassette by homologous recombination in EC7.1 embryonic stem (ES) cells. Clones were screened for the homologous recombination event by long-range PCR. The PCR products were digested with specific enzymes (EcoRV for the left arm and ScaI for the right arm) to confirm the identities. (B) Cloning vectors for the generation of the recombination cassette. pF-WLPLF3: a transgene could be cloned at the Apal or BamHI site in a way that the downstream WPRE-pA will allow proper transcription. The floxed PGK-puro selection marker could be removed from the targeted allele after recombination if so desired. pFLSL-WF3: a transgene could be cloned at the NotI site which is downstream of a floxed-STOP sequence (a tandem array of four SV40 polyA signals). The transgene will express once the STOP sequence is deleted by Cre-recombinase.
To facilitate the cloning of the transgene, we made two exchange vectors; one for constitutive expression (pF-WLPLF3), and another for conditional expression, mediated by the cre/loxP system (pFLSL-WF3) (Figure 1B). A woodchuck hepatitis post-transcriptional regulatory element (WPRE) was also incorporated to enhance transgene expression. Both vectors have the recombination sites FRT and F3 flanking the transgene cassette, and in the case of pFLSL-WF3, a floxed-STOP cassette was inserted upstream of the transgene to prevent expression until a cre-mediated deletion of the STOP cassette had taken place, allowing spatial and temporal control of expression4,5.
The 1F1 ES cells were cultured with primary mouse embryonic fibroblasts (PMEF) (Millipore, Billerica, MA) as feeders in KnockOut DMEM (Life Technologies, Carlsbad, CA) supplemented with 15% FBS (Tissue Culture Biologicals, Los Alamitos, CA) 1,000 U/ml LIF (ESGRO) (Millipore, Billerica, MA), 100 µM non-essential amino acids (Life Technologies, Carlsbad, CA), 2 mM GlutaMAX (Life Technologies, Carlsbad, CA), 55 µM 2-mercaptoethanol (Life Technologies, Carlsbad, CA), and 25 U Pen/Strep (Life Technologies, Carlsbad, CA), at 37°C and 5% CO2. They were passaged one-tenth when confluent, typically every two days. To illustrate the efficiency of RMCE in knocking-in and expressing a green fluorescent protein (GFP) marker, we transfected 5 x 106 1F1 ES cells by electroporation with 15 µg of pF-GFP-WLPLF3 and 15 µg of pCAG-Flpe6 (Addgene plasmid 13787). Electroporation was carried out in a 4 mm cuvette on the Gene Pulser Xcell (Bio-Rad, Hercules, CA) at 250 volts and 500 µF capacitance. The transformants were plated onto three 100 mm dishes containing DR4 mouse fibroblasts as feeders (Applied Stem Cell, Menlo Park, CA); puromycin (1 µg/ml) (Sigma-Aldrich, St. Louis, MO) was applied 24 hr post-plating. Resistant clones were allowed to grow for seven days after which a total of 96 individual colonies were hand picked, trypsinized, and cultured in a 96-well plate. We made replica plates when the colonies were confluent so that one set of cells was cryo-preserved in 10% DMSO (Sigma-Aldrich, St. Louis, MO) at -80°C, one set was subjected to a G418 (250 µg/ml) (Life Technologies, Carlsbad, CA) sensitivity test, and another set of cells was lysed to isolate genomic DNA for genotyping analysis7. From the 96 clones tested, we found 8 clones that were G418 sensitive (Figure 2B), indicating loss of the original neo cassette, through the exchange of the transgene cassette (Figure 2A). The clones that were puromycin resistant but still retained G418 resistance should be clones that had random integration of the transgene cassette without displacing the neo cassette at the ROSA26 locus. The exchange was further confirmed by PCR analysis (Figure 2C). Briefly, a pair of primers annealing to the upstream sequence of the ROSA26 locus and the GFP (arrows indicated in Figure 2A) were employed to amplify the cassette exchange product. We then analyzed GFP expression of the knock-in as well as a randomly picked background clone (PuroR G418R). By using fluorescent microscopy on a Axiovert 200M microscope (Zeiss, Jena, Germany) on live ES cells, we confirmed two of the selected knock-in clones (B7 and B12) exhibited GFP expression while the background clone D5 did not (Figure 2D), suggesting that the knock-in GFP marker at the ROSA26 locus was functionally expressed.
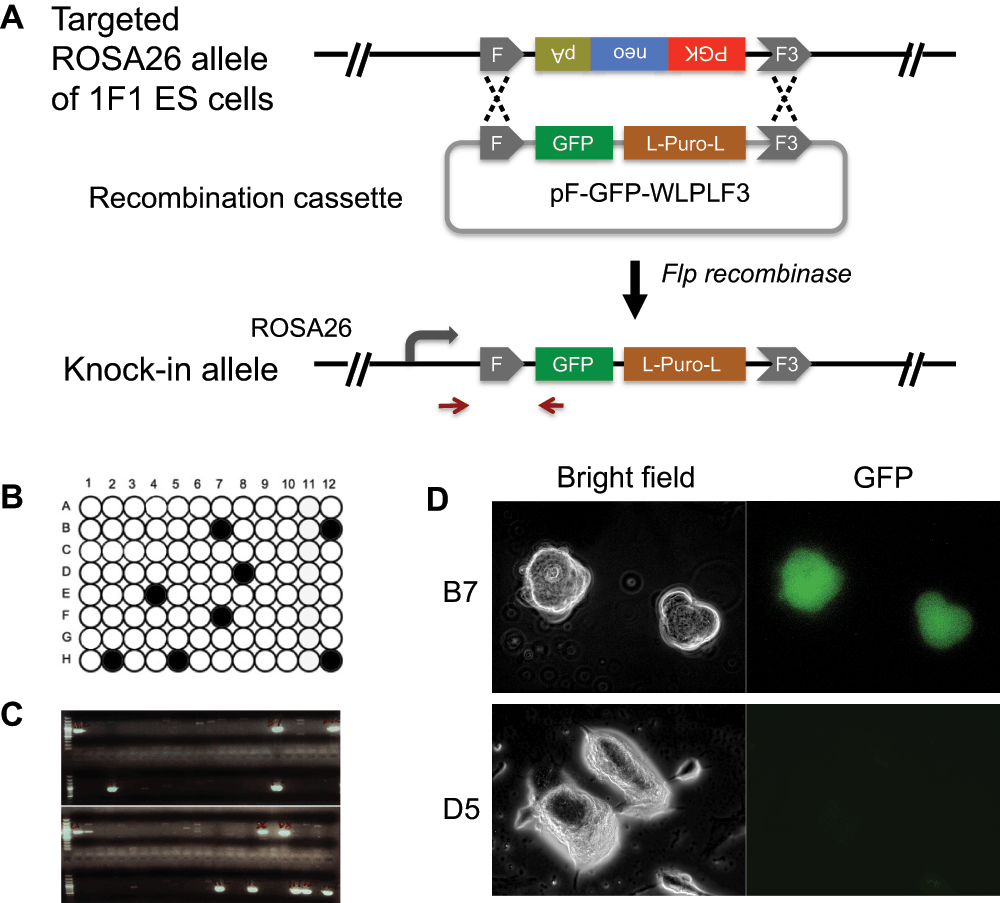
(A) Schematic of RMCE. Circular plasmids of the exchange vector pF-GFP-WLPLF3 and the pCAG-Flp expression vector were transfected into 1F1 embryonic stem (ES) cells. Recombinants were selected for puromycin resistance; each individual clone was then seeded onto the well of a 96-well plate. (B) G418 sensitivity test. The black wells represent those ES cell clones that were killed by G418 (PuroR G418S), indicating the loss of the originally tagged neo marker. (C) Genotyping analysis for the RMCE event. ES cells were analyzed by PCR (the primers are represented by the arrows in Figure 2A) for the correct integration of GFP into the ROSA26 allele. (D) Expression of GFP. Knock-in ES cell clone B7 is expressing GFP as observed live under a fluorescent microscope. The background clone D5 (PuroR G418R) that has a random integration does not have GFP expressed.
In summary, we present here a protocol and the associated reagents required for a very efficient gene knock-in at the ROSA26 locus in mouse ES cells using recombinase-mediated cassette exchange. Two cloning vectors are described for the generation of a transgene exchange cassette in a single cloning step. The timeline from transfection of ES cells to the isolation of recombinant clones is approximately two to three weeks. We observed that the recombination rate of RMCE was quite high so that recombinants could be isolated easily. For the GFP transgene we observed an 8% recombination rate. Ninety-six clones of two other RMCE transgenesis (Pik3r1 and NpHR-eYFP) generated by the same approach were screened (by PCR and the G418 sensitivity test) as described above, yielding 12% and 31% recombination rates, respectively, suggesting that no more than a single 96-well plate of clones need to be harvested and analyzed. Moreover, the screening of recombinants was straightforward and easy to interpret. Unlike gene targeting by homologous recombination, the PCR genotyping for RMCE events in these experiments was very efficient because of the small size of amplicons (~500 bp). As a side note, we observed that PCR was optimal when using DMSO (2.5%) and a reduced Mg2+ concentration (1 mM) in the PCR buffer, an observation we encountered whenever we attempted to amplify at the ROSA26 locus. In parallel, the G418 sensitivity test served as confirmatory evidence of gene exchange. Overall, we found that the experimental procedure is simple to perform and exhibits high efficiency. It could easily be adopted by other laboratories. The 1F1 ES cells were derived from a F1 hybrid strain with a background of C57BL/6 and 129X1/SvJ. Our experience with these hybrid ES cells is that they contribute to the germline efficiently, even after a variety of in vitro manipulations3,8. The FRT-neo-F3 cassette was targeted to the C57BL/6 allele into which the transgene will be exchanged; mice derived could then be backcrossed to attain a pure C57BL/6 background which, depending on the intended mouse study, might be desirable.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)