Introduction
Inhaled glucocorticoids are the first-line treatment for asthma1–3. Glucocorticoids bind to the glucocorticoid receptor to reduce the expression of genes that produce a variety of pro-inflammatory mediators and mucus in the lung4–6. The most commonly prescribed glucocorticoids are beclomethasone dipropionate (BDP), triamcinolone acetonide (TCL), budesonide (BUD), fluticasone propionate (FLT), and flunisolide (FLN)1. BDP is a pro-drug and requires removal of the C-21 propionate group to become pharmacologically active; the active drug is beclomethasone 17-monopropionate, referred to as [M1] (Figure 1)7. Pharmacological inactivation and clearance of glucocorticoids, such as BDP and its active metabolite [M1], is mediated, in part, by cytochrome P450 (CYP) enzymes (Figure 1).
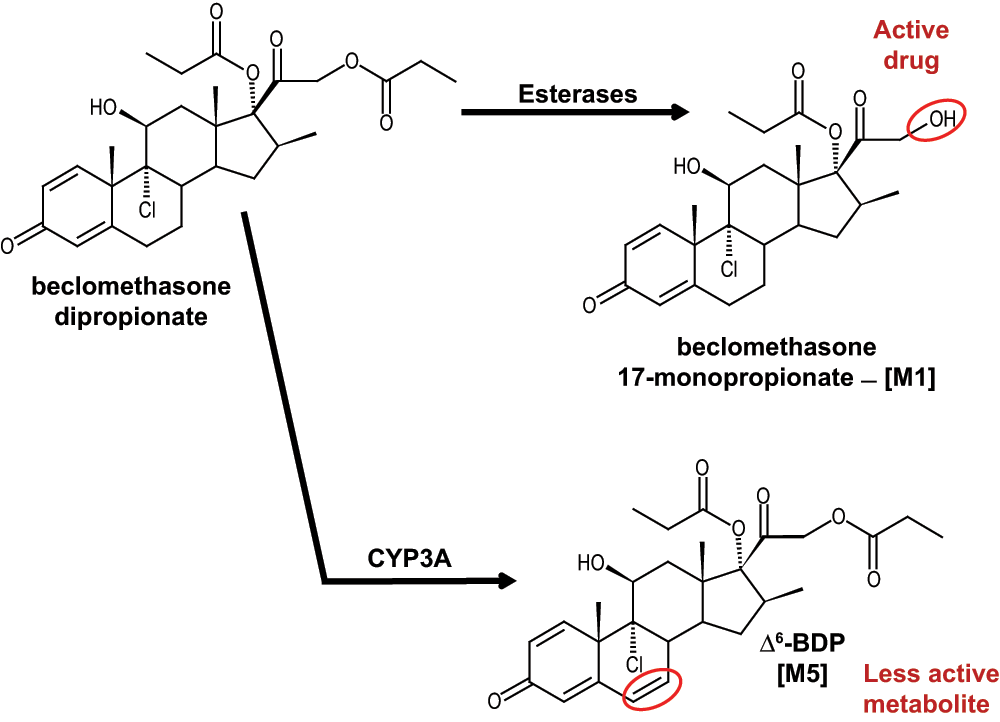
Figure 1. Metabolic scheme for the production of [M1] (the active form of the drug) by esterase enzymes and [M5] by CYP3A enzymes.
In humans, CYP3A4, 3A5, and 3A7 are the primary CYP enzymes involved in glucocorticoid metabolism8–11. CYP3A4 is the most abundant CYP3A enzyme in the liver and intestines8,12,13, CYP3A5 is more prevalent in the lung than the liver12,14–16, and CYP3A7 is expressed in fetal liver, but diminishes after birth when CYP3A4 becomes the dominant adult hepatic CYP3A enzyme17,18. Expression of CYP3A7 in fetal and adult respiratory tissue has also been observed16.
Regulation of CYP3A enzymes in response to glucocorticoid treatment has been extensively characterized in the liver, but less is known about this phenomenon in the lung. In hepatocytes, CYP3A enzyme induction is mediated by the pregnane X receptor (PXR)19,20 (Figure 2A). However, PXR is not expressed in the lung21. Glucocorticoids can also influence CYP3A induction via the glucocorticoid receptor (GR) and the constitutive androstane receptor (CAR) in the liver22,23. Briefly, glucocorticoids bind GR in the cytosol, which forms a homodimer and translocates into the nucleus, leading to increased transcription of CAR. CAR forms a heterodimer with the retinoid X receptor alpha (RXRα), which binds to the RXR-response element and induces the expression of CYP3A enzymes (Figure 2A)22. Previous work by Hukannen et al. demonstrated that CAR was not expressed in A549 (adenocarcinomic human alveolar basal epithelial) cells and suggested that glucocorticoid binding to GR may directly regulate CYP3A gene expression in A549 cells (Figure 2B), based on inhibition using RU-48615,24. However, these pathways have not been evaluated in primary lung cell cultures or lung tissue.
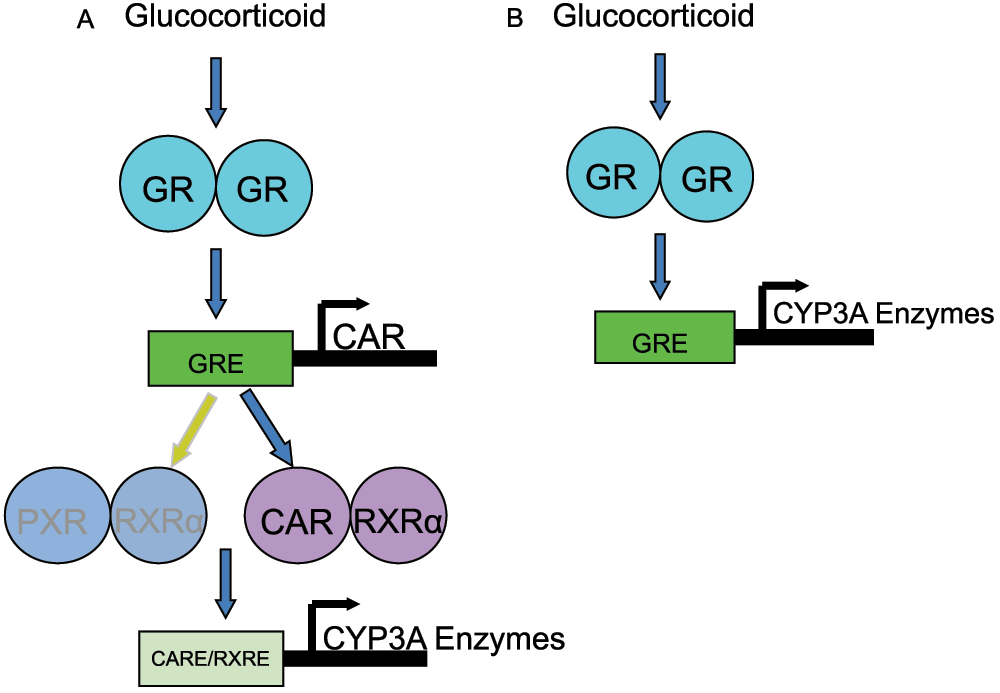
Figure 2. Possible mechanisms for the induction of CYP3A genes in lung cells.
(A) Active glucocorticoid will bind to the glucocorticoid receptor (GR), which forms a homodimer and translocates to the nucleus. The homodimer binds to its response element (GRE) and induces the expression of either the pregnane X receptor (PXR) or the constitutive androstane receptor (CAR). CAR or PXR (though this receptor is not expressed in the lungs) forms a heterodimer with the retinoic X receptor alpha (RXRα) which in turn induces the expression of the CYP3A enzymes via binding of the respective response-elements (CARE and/or PXRE). (B) Active glucocorticoid will bind to the glucocorticoid receptor (GR), which forms a homodimer and translocates into the nucleus. The homodimer binds to its response element (GRE) and induces the expression of CYP3A enzymes.
The purpose of this study was three fold: to evaluate the changes in the expression of CYP3A mRNA in primary lung cells treated with glucocorticoids; to determine which pathway was responsible for glucocorticoid-induced changes in CYP3A mRNA expression; and to determine the role of BDP metabolism in this phenomenon. The cell lines used in this study were BEAS-2B (immortalized bronchial epithelial cell line), NHBE (normal human bronchial/tracheal epithelial cells), lobar epithelial cells (secondary bronchus epithelial cells), primary cells recovered from tracheal washes of pediatric patients on ventilation, SAEC (small airway epithelial cells), and A549 (human lung adenocarcinoma) cells. It was hypothesized that CYP3A5 mRNA induction in primary cells by BDP11 and other glucocorticoids would occur via a mechanism involving GR/CAR/RXRα, as previously demonstrated using hepatocytes.
Methods
Chemicals, reagents, and treatments
Beclomethasone dipropionate (BDP), triamcinolone acetonide (TCL), fluticasone propionate (FLT), flunisolide (FLN), budesonide (BUD), prednisolone, ammonium acetate, eserine, and methanol were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO). Paraoxon was purchased from Chem Service (West Chester, PA).
Cell culture
A549 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's Modified Eagle Medium (DMEM) fortified with 5% fetal bovine serum (Life Technologies, Grand Island, NY). SAEC cells (LONZA, Walkersville, MD; donor numbers 11662, 14453, 14457) were cultured in small airway epithelial growth medium, supplemented with the SAGM bullet kit (LONZA). Cells were cultured with and without hydrocortisone by adding or not adding the hydrocortisone component from the SAGM bullet kit. NHBE cells (LONZA; donor numbers 15268, 5S03795) were grown in bronchial epithelial cell growth medium (BEGM Bullet kit) (LONZA). BEAS-2B cells (American Type Culture Collection) were cultured in LHC-9 medium (Life Technologies). Lobar cells (donor number 01334) were cultured in BronchiaLife Basal Medium supplemented with the BronchiaLife B/T supplement kit (Lifeline Cell Technology, Walkersville, MD). All cells except A549 cells were plated in 12-well plates pre-coated with LHC basal medium (Life Technologies) and cultured in the presence of hydrocortisone. Tracheal epithelial cells were recovered from tracheal washes from mechanically ventilated pediatric patients in the neonatal intensive care unit and pediatric intensive care unit at Primary Children’s Medical Center at the University of Utah, with IRB approval (00026839). Briefly, cells were separated from sputum by centrifugation at 900 x g for 30 min in 14 mL of DMEM/F12 media. Cells were plated in a 12-well plate pre-coated with 2% gelatin (Life Technologies) and cultured in DMEM/F12 media + 10% fetal bovine serum (FBS) (Life Technologies). All cells were cultured in an atmosphere of 5% CO2:95% air at 37ºC.
Cell treatments
Cell treatments were prepared in treatment media with a final concentration of DMSO less than 1%. Cells were treated at ~70% confluence. A549 cells were cultured in OPTIMEM (Life Technologies) and SAEC cells were cultured in growth media either with or without hydrocortisone and with or without heat inactivated and/or charcoal-stripped FBS. All other cell lines were treated in their respective growth medium, and heat inactivated to eliminate esterase activity from the FBS, which would metabolize BDP before it could diffuse into the cells. Cytotoxicity assays were performed using the Dojindo Cell Counting Kit-8 (Dojindo Laboratories, Rockville, MD) to determine glucocorticoid, esterase inhibitor, and ketoconazole concentrations exhibiting <20% cytotoxicity in A549 cells. All other cell lines were treated with the same concentrations as determined with A549 cells. Glucocorticoid treatments were as follows: BDP (10 μM), TCL (1 μM), BUD (10 μM), FLT (1 μM), and FLN (100 nM). Pre-treatments in various experiments included ketoconazole (Sigma-Aldrich Chemical Company) (50 μM, 10 μM, and 1 μM, to antagonize GR), esterase inhibitors (1:1 mixture of eserine and paraoxon, each at 175 μM, to inhibit [M1] formation), and 1-aminobenzotriazole (Sigma-Aldrich Chemical Company) (1-ABT; 200 μM, to inhibit P450-mediated metabolism) for 2 h prior to a 22 h glucocorticoid co-treatment. Controls were treated with an equivalent concentration of DMSO. All A549 cell treatments were carried out in 6-well plates for 24 h (n=6). All other cell lines were cultured in pre-coated 12-well plates and treated for 24 h (n=3).
Analysis of BDP metabolites
After treatment, BDP and its metabolites were extracted from the collected media by adding 2x volume (6 mL for A549, 4 mL for all other cell lines) methyl tert-butyl ether containing 1 nM prednisolone (internal standard for quantification) and shaking for 25 min. Samples were clarified by centrifugation, the organic fraction was collected, dried under air, reconstituted in 100 µL 1:1 H2O:MeOH, clarified again by centrifugation, and transferred to autosampler vials for analysis by liquid chromatography-mass spectrometry (LC/MS/MS). LC/MS/MS was conducted on a Thermo LCQ Advantage Max ion trap instrument equipped with a Finnigan Surveyor LC pump, Surveyor Autosampler and universal Ion Max source operated with Thermo Xcalibur software version 2.0 (Thermo Fisher Scientific, Waltham, MA) as previously described11.
Quantitative reverse transcription-PCR
Total RNA was isolated from cells using TRIzol reagent (Life Technologies). cDNA was synthesized using iScript Reverse Transcription Supermix for qPCR (BIO RAD, Hercules, CA). qPCR was performed using either LightCycler 480 Probes Master mix (CYP3A5*1) or LightCycler 480 SYBR Green I Master Mix (all other genes) (Roche, Indianapolis, IN) with a LightCycler 480 System. The PCR program for the probe mix consisted of a 5 min incubation at 95ºC, followed by 45 cycles of 95ºC for 10s, 55ºC for 30s, then 72ºC for 1s. The PCR program for SYBR Green I mix consisted of a 5 min incubation at 95ºC, followed by 40 cycles of 95ºC for 10s, 63ºC for 5s for CYP3A4, CYP3A7 and β2-microglobulin. For GR and CAR, annealing was performed at 65ºC for 5s and extension at 72ºC for 10s. mRNA copy number was determined from standard curves for each gene and was normalized using β2-microglobulin. Primer sequences for the various genes are listed in Table 125.
Table 1. Primer sequences for qPCR assays.
| CYP3A5*1 | F-5´ CCTATCGTCAGGGTCTCTGGAA 3´
R-5´ TGATGGCCAGCACAGGGA 3´
Probe [6FAM]ATGTGGGGAACGTATGAA[BHQ1] |
| CYP3A5-all | F-5´ CGTCAGGGTCTCTGGAAATTTG 3´
R-5´ CACGTCGGGATCTGTGATGG 3´ |
| CYP3A4 | F-5´ GAAAGTCGCCTCGAAGATAC 3´
R-5´ ACGAGCTCCAGATCGGACAG 3´ |
| CYP3A7 | F-5´ TTCCGTAAGGGCTATTGGAC 3´
R-5´ TCTGTGATAGCCAGCATAGG 3´ |
Glucocorticoid
receptor | F-5´ CCAACGGTGGCAATGTGAAA 3´
R-5´ CCGCCAGAGGAGAAAGCAAA 3´ |
Constitutive
androstane
receptor | F-5´ CCGTGTGGGGTTCCAGGTAG 3´
R-5´ CAGCCAGCAGGCCTACGAAC 3´ |
β2-
microglobulin | F-5´ GATGAGTATGCCTGCCGTGTG 3´
R-5´ CAATCCAAATGCGGCATCT 3´ |
siRNA-mediated protein knockdown
Pre-annealed, short interfering “Smart Pool” siRNAs specific to human GR were purchased from Dharmacon (Waltham, MA). siRNA directed against GFP (negative control)26 was synthesized at the University of Utah oligonucleotide synthesis core and annealed by combining 40 µM of each strand and incubating in annealing buffer (100 mM potassium acetate, 30 mM HEPES KOH, 2 mM magnesium acetate adjusted to pH 7.4) for 1 min at 90ºC followed by 1 h at 37ºC, in a final volume of 0.5 mL. A549 cells were plated into 6-well plates containing 20 nM siRNA per well, previously complexed with Lipofectamine 2000 (Life Technologies) using a ratio of 3:2 lipid to siRNA in 100 µL of OPTIMEM (Life Technologies). The cells were grown for 48, 72, and 96 h to determine the time at which maximum decreases in GR mRNA occurred (72 h). In subsequent experiments, cells were treated with DMSO, 10 µM BDP, or 10 μM BDP + 175 µM esterase inhibitors (1:1 eserine:paraoxon) for 24 h to determine the effects of attenuated GR expression on the induction of CYP3A5 in A549 cells.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4.02 software for Windows (San Diego, CA). One-way ANOVA and Dunnett’s post-hoc test were used with p<0.05. All data are represented as a mean with error bars representing standard deviation.
Results
Inhibition of [M1] formation prevented CYP3A5 mRNA induction by BDP in A549 cells
Media from A549 cells treated with BDP (10 µM) for 24 h was extracted and analyzed for metabolites of BDP produced by CYP3A enzymes. The only CYP3A-mediated metabolite detected was [M5] (Figure 1 and Figure 3A)11. For the remainder of the studies, [M1], the active metabolite, was used as a marker for esterase activity and [M5] was used as a marker for CYP3A5 activity. Only CYP3A5*1 mRNA was detected in A549 cells. CYP3A4 and CYP3A7 mRNA were not detected in A549 cells, as previously documented11,16. BDP treatment significantly induced the expression of CYP3A5 mRNA (~2-fold) compared to the DMSO control (Figure 3B). Inhibiting the production of [M1] using esterase inhibitors also blocked the induction of CYP3A5 mRNA (Figure 2A and Figure 2B); esterase inhibitor (EI) treatment alone had no effect on CYP3A5 expression. 1-ABT, a mechanism-based inactivator of P450 enzymes, also inhibited esterase activity (i.e. [M1] formation) (Figure 3A), and as a result, prevented the induction of CYP3A5 mRNA (Figure 3B). The mechanism by which 1-ABT inhibits esterases is not known.
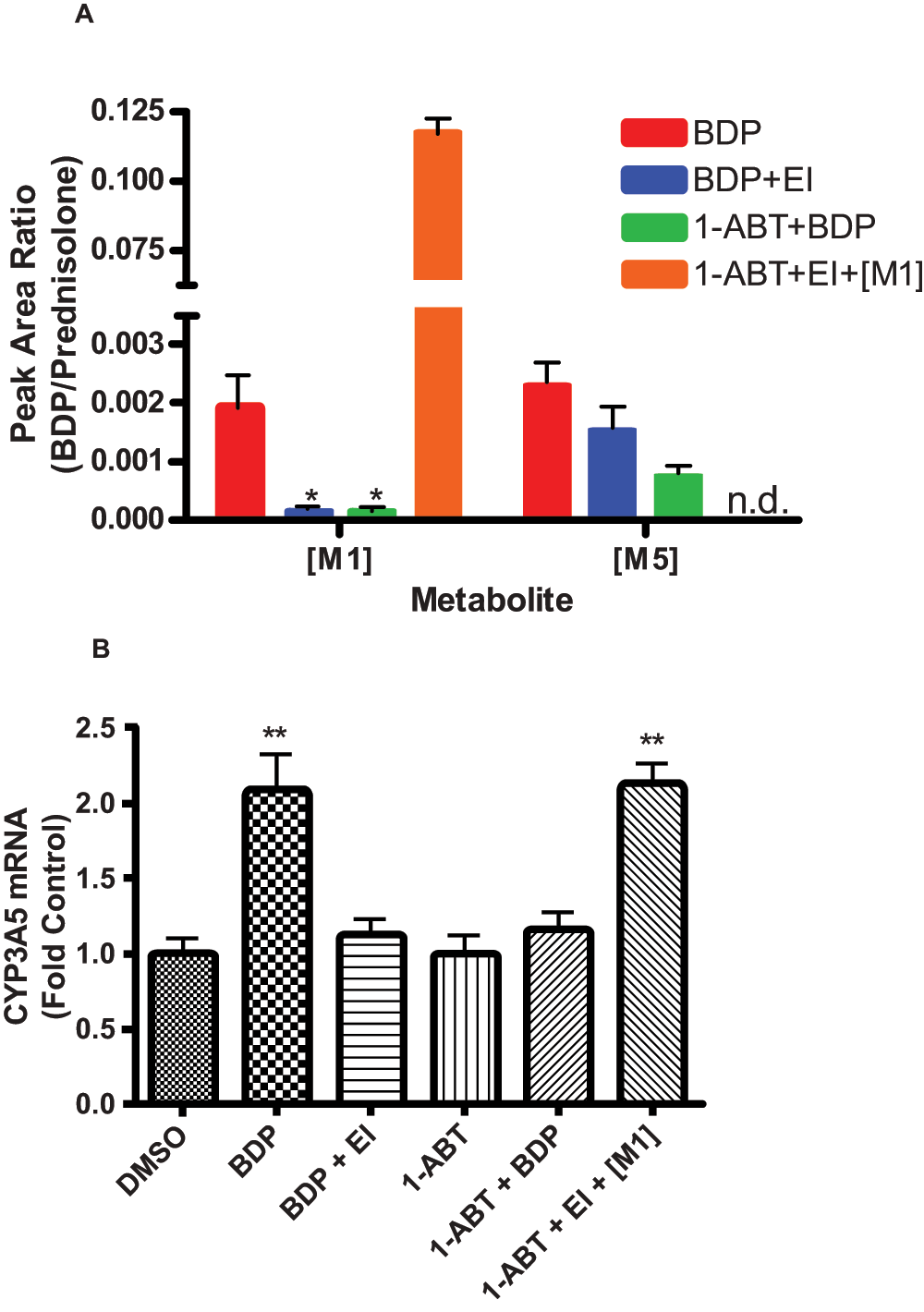
Figure 3. [M1] is required to induce CYP3A5 mRNA.
(A) Relative quantities of [M1] and [M5] measured by LC/MS/MS in A549 cell culture media following beclomethasone dipropionate (BDP) treatment alone, BDP + esterase inhibitors (EI), 1-aminobenzotriazole (1-ABT) + BDP, and 1-ABT + EI + [M1]. (B) CYP3A5 mRNA detected in A549 cells following DMSO control, BDP treatment alone, BDP + EI, 1-ABT alone, 1-ABT + BDP, and 1-ABT + EI + [M1]. Values are expressed as fold over DMSO controls. Statistics used for data analysis were one-way ANOVA with Dunnett’s post-hoc test. Data are the mean and standard deviation from n=6 replicates. * p<0.05, ** p<0.01.
[M1] was sufficient to induce CYP3A5 mRNA in A549 cells
Cells were treated with [M1] in either the absence or presence of 1-ABT and esterase inhibitors. [M1] treatment was sufficient to induce CYP3A5 mRNA (~2-fold), without the requirement of esterases to produce [M1] (Figure 3B), indicating that CYP3A5 mRNA induction in A549 cells was mediated by [M1].
GR, but not CAR, regulated the induction of CYP3A5 mRNA in A549 cells
GR and CAR mRNA were quantified in A549 cells. A significant increase in GR mRNA (~2.5-fold) was observed following 24 h treatment with BDP (Table 2), consistent with previous studies15, suggesting that GR, not CAR, was responsible for the induction of CYP3A5 message in A549 cells.
Table 2. Comparison of glucocorticoid receptor (GR), constitutive androstane receptor (CAR), and CYP3A5 mRNA expression in lung cell cultures.
| Cell type | GR
expression | GR induction by
GC treatment | CAR
expression | CAR induction
by GC treatment | CYP3A5
mRNA | CYP3A5 induction
by GC treatment |
|---|
| Beas-2B | + | N.D. | N.D. | N.D. | N.D. | N.D. |
| NHBE | + | N.D. | N.D. | N.D. | N.D. | N.D. |
Patient
tracheal
washes | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Lobar | + | N.D. | N.D. | N.D. | N.D. | N.D. |
| A549 | + | 2.4 ± 0.35 ** | N.D. | N.D. | + | 2.1 ± 0.55 ** |
| SAEC | + | N.D. | N.D. | N.D. | 1 out of 3
patients | N.D. |
Inhibition of GR with ketoconazole attenuated CYP3A5 mRNA induction by glucocorticoids in A549 cells
Ketoconazole is a competitive antagonist of GR25. Ketoconazole alone had no significant effect on CYP3A5 mRNA expression as compared to DMSO controls. As the concentration of ketoconazole was decreased, dose-dependent increases in the expression of CYP3A5 mRNA were observed for BDP, TCL, FLT, BUD, and FLN (Figure 4A–E): BDP caused a ~2-fold induction, BUD caused a ~4-fold induction, TCL caused a ~5.5-fold induction, FLT caused a ~3.5-fold induction, and FLN caused a ~5.5-fold induction, relative to their respective controls. These data confirm the hypothesis that the induction of CYP3A5 mRNA in A549 cells was mediated by GR. BDP or FLT paired with KTZ 1 µM treatment also showed further induction of CYP3A5 mRNA as compared to controls (~3.5 for BDP and ~6.5 for FLT). However, the basis and significance for this enhanced induction are not clear at this time.
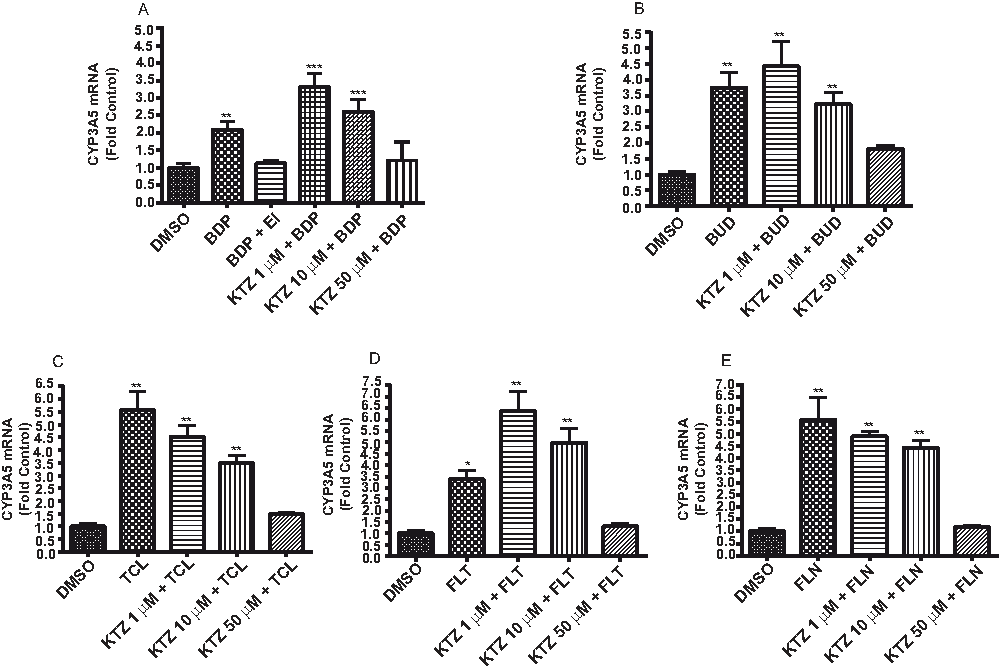
Figure 4. Ketoconazole inhibits the induction of CYP3A5 through the glucocorticoid receptor (GR).
CYP3A5 mRNA detected in A549 cells treated with (A) beclomethasone dipropionate (BDP), (B) budesonide (BUD), (C) triamcinolone acetonide (TCL), (D) fluticasone propionate (FLT), and (E) flunisolide (FLN), with and without ketoconazole (KTZ), a competitive antagonist for GR. Results are presented as fold over DMSO controls. Statistics used for data analysis were one-way ANOVA with Dunnett’s post-hoc test. Data are the mean and standard deviation from n=6 replicates. * p<0.05, ** p<0.01, *** p<0.001.
siRNA-mediated knockdown of GR also attenuated CYP3A5 mRNA induction by BDP in A549 cells
Cells were transfected with siRNA and grown for 48, 72, and 96 h to determine the time of maximum GR mRNA knock down (Figure 5A). Maximum suppression occurred as early as 48 h, but the 72 h time point was chosen for further experiments to ensure efficient GR protein depletion. An approximate 2-fold induction of CYP3A5 mRNA was observed in A549 cells following treatment with BDP in control cells transfected with “nonsense” siRNA directed against GFP. Consistent with previous results (Figure 3A and Figure 3B), CYP3A5 mRNA induction was prevented by esterase inhibitors (Figure 5B). Cells transfected with siRNA targeted for GR mRNA showed no change in CYP3A5 mRNA with BDP treatment (Figure 5B), further confirming the role of GR in directly regulating the induction of CYP3A5 mRNA in A549 cells treated with BDP and presumably the other glucocorticoids used in Figure 4.
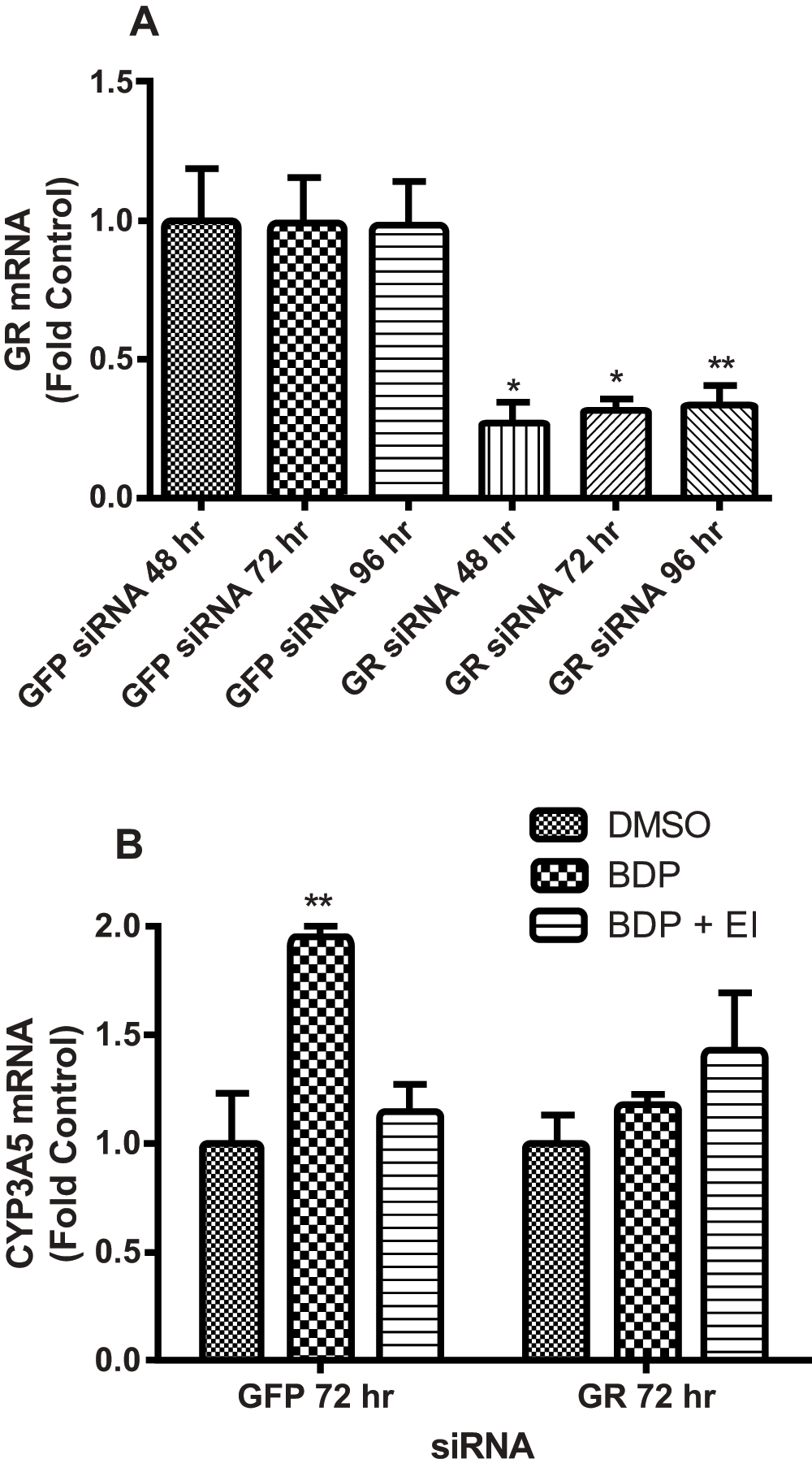
Figure 5. Glucocorticoid receptor (GR) siRNA blocks the induction of CYP3A5 by beclomethasone dipropionate (BDP).
(A) siRNA-mediated GR knockdown in A549 cells at 48, 72, 96 h compared to "nonsense" GFP siRNA (negative control), expressed as fold over designated GFP control for each time point. (B) Cells were exposed to siRNA for 72 h then treated with DMSO, BDP, or BDP + esterase inhibitor (EI). Statistics used for data analysis were one-way ANOVA with Dunnett’s post-hoc test. Data are the mean and standard deviation from three replicates. * p<0.05, ** p<0.01.
CYP3A5 was not expressed or induced by glucocorticoid treatment in tracheal/bronchial epithelial cells
Neither CYP3A5*1 mRNA nor any other variant form of CYP3A5 mRNA was detected or induced by glucocorticoids in NHBE, BEAS-2B, lobar, and freshly isolated tracheal wash samples (Table 2).
SAEC cells expressed CYP3A5, but mRNA for CYP3A5 was not induced by glucocorticoid treatment
SAEC cells from three separate donors were evaluated for CYP3A5*1 and other variant forms of CYP3A5 mRNA expression and induction in response to glucocorticoid treatment. Initial experiments demonstrated that mRNA for CYP3A5*1, but not CYP3A4 or 3A7, was expressed in one of the three SAEC samples (donor number 11662), but that expression levels were not altered by glucocorticoid treatment. It was hypothesized that the high concentration of hydrocortisone (500 µM) in the SAEC growth media prevented the induction of CYP3A5 mRNA by substantially lower concentrations of the glucocorticoids used in the treatments. Elimination of hydrocortisone from the media decreased the basal expression of CYP3A5 mRNA (Figure 6). However, no change in mRNA abundance was observed over a 24 h treatment period with BDP. Furthermore, neither increasing the treatment concentration of BDP to 50 µM, nor treatment with [M1] at 150 µM led to an increase in CYP3A5 mRNA in SAEC cells. It was subsequently hypothesized that phthalates or other substances in the FBS might alter GR function and CYP3A5 mRNA induction by glucocorticoids27. However, neither heat inactivation nor charcoal-stripping of the FBS in media with or without hydrocortisone led to CYP3A5 mRNA induction. The various manipulations to SAEC culture conditions and results for CYP3A5 induction are summarized in Table 3.
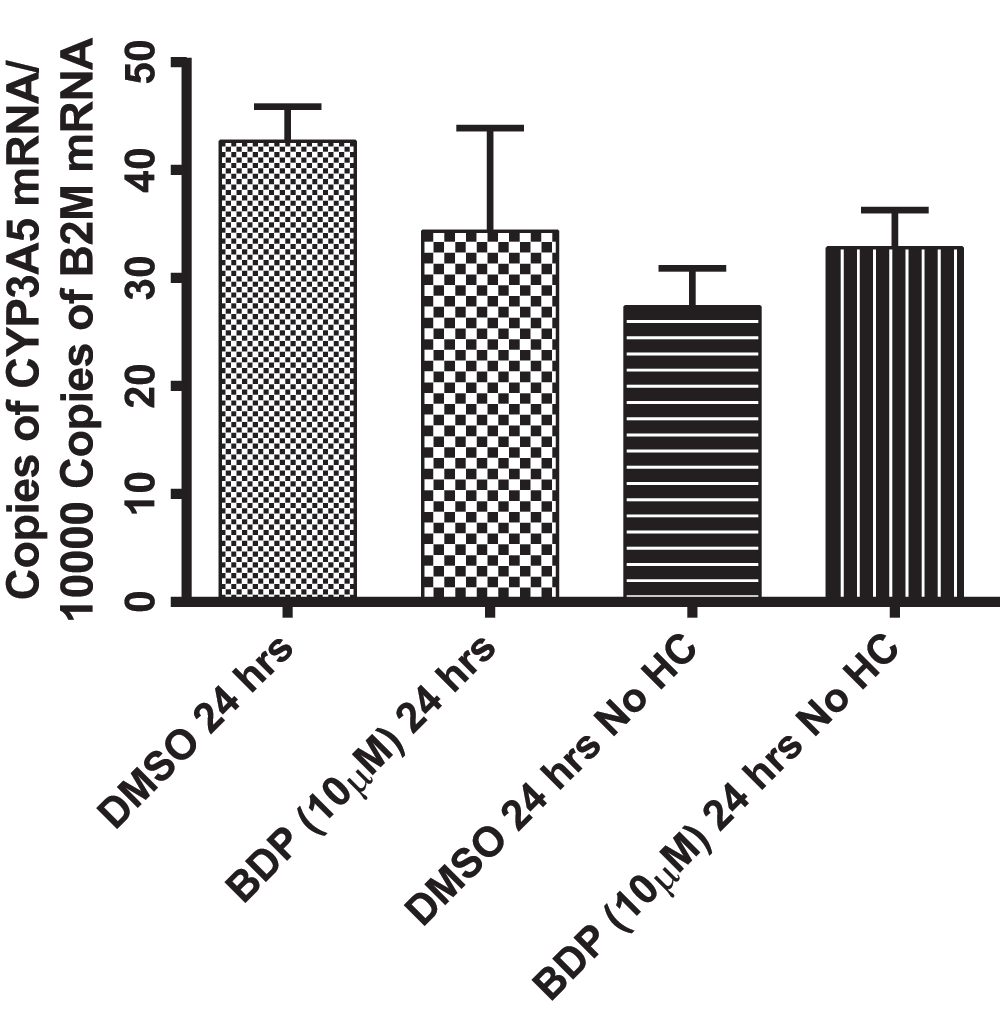
Figure 6. Beclomethasone dipropionate (BDP) treatment in SAEC cells results in no change in CYP3A5 mRNA.
SAEC cells treated with BDP (10 µM; 24 h) or DMSO matching controls with and without hydrocortisone (HC). There was no significant difference between treatments or matching controls using one-way ANOVA with p<0.05.
Table 3. Modifications made to SAEC culture media.
| Basal culture conditions | Experimental modifications | Result |
|---|
| Cultured in growth media | -Heat inactivated media for treatment
-10 µM BDP for 24 h | -Basal CYP3A5 mRNA expression observed
in 1 out of 3 patients
-No change in CYP3A5 mRNA |
Cultured in growth media without
hydrocortisone | -Heat inactivated media for treatment
-10 µM BDP for 24 h | -Lowered basal level of CYP3A5 mRNA
-No change in CYP3A5 mRNA |
Cultured in growth media without
hydrocortisone | -Heat inactivated media for treatment
-50 µM BDP or 105 µM [M1] for 24 h | -No change in CYP3A5 mRNA |
Cultured in growth media without
hydrocortisone | -Treated in heat inactivated and charcoal
stripped FBS
-50 µM BDP or 105 µM [M1] for 24 h | -No change in CYP3A5 mRNA |
Discussion
Inhaled glucocorticoids are used to control undesirable symptoms in asthmatic patients. However, about 30% of the population does not benefit from this first-line treatment6. Prior work demonstrated that the five most commonly prescribed glucocorticoids used in the treatment of asthma are metabolized by CYP3A enzymes, specifically CYP3A4, CYP3A5, and CYP3A710,11. Therefore, it has been proposed that unusually high rates of metabolism of glucocorticoids in lung cells by these enzymes might contribute to the decrease and/or lack of efficacy in some individuals. However, it is not understood how the expression of CYP3A enzymes is regulated in the lung in response to glucocorticoid treatment, despite extensive knowledge of this phenomenon in hepatocytes and the liver22.
Using A549 cells, it was demonstrated that CYP3A5*1 mRNA was induced by glucocorticoid treatment (Figure 3B and Figure 4A–E); neither CYP3A4 nor CYP3A7 mRNA were detected in A549 cells. Subsequent studies using a competitive antagonist of GR (ketoconazole) and siRNA selective for GR mRNA, demonstrated that inhibition of GR function prevented the induction of CYP3A5 mRNA by BDP and other glucocorticoids in A549 cells (Figure 4A–E and Figure 5B). It was also demonstrated that CAR mRNA was not expressed by lung cells, consistent with previous data15, and therefore could not be involved in the regulation of CYP3A5 expression by glucocorticoids as occurs in hepatocytes. It was concluded that CYP3A5 expression was directly regulated by GR (Figure 2B). Schuetz et al.24 previously described two “half sites” of GR (TGTTCT) separated by 160 bp in the promoter region of CYP3A5 in HepG2 cells and in human and rat hepatocytes. It was demonstrated that dexamethasone induced the expression of CYP3A5 by the GR homodimer binding to these two joined “half-sites” which could be blocked by RU-486, a GR antagonist. It is plausible these same sites are involved in the regulation of CYP3A5 in lung cells by BDP and other glucocorticoids.
Regardless of the exact mechanism of regulation, the current results illustrate that glucocorticoids have the capacity to induce the expression of CYP3A5 in A549 cells. These data, in conjunction with prior metabolism studies of glucocorticoids10,11, support the hypothesis that treating patients with glucocorticoids could increase levels of CYP3A5 in the lung, and therefore increase pulmonary glucocorticoid metabolism, ultimately increasing clearance, and potentially decreasing the concentration of active drug in lung cells. Though most of the population expresses the inactive form of CYP3A5 (CYP3A5*3)13,28 those expressing CYP3A5*1, the active form of CYP3A513, would exhibit increased clearance of the drug, and therefore could account for at least some of the 30% of patients who do not respond to inhaled glucocorticoid therapy.
In order to further support the hypothetical scenario above, the induction of CYP3A enzymes by glucocorticoids in various lung cells was studied. CYP3A5 mRNA expression was quantified in primary lung cells, which presumably more closely model epithelial cells of the human respiratory tract and lung. NHBE, lobar, and cells recovered from tracheal washes of mechanically ventilated children were evaluated for CYP3A enzyme expression and induction by glucocorticoids. Results in Table 2 show that CYP3A mRNA was not expressed in cells of the conducting airways in response to glucocorticoid treatment, indicating that these epithelial cells likely do not play a role in CYP3A-dependent metabolism of glucocorticoids in the lung. In contrast, select donor samples of SAEC cells, representing cells of the distal bronchioles, alveolar ducts, and alveoli, did express CYP3A5 (Table 2). However, there was no change in CYP3A5 mRNA when these cells were treated with glucocorticoids. A thorough examination of potential confounding issues associated with cell culture revealed a high concentration of hydrocortisone (500 µM) in the growth media. Because cells were treated with only 10 µM BDP, it would stand to reason that no change in CYP3A5 mRNA would occur because CYP3A5 expression would already be maximized as a result of hydrocortisone activating the GR pathway.
Experiments conducted in A549 cells showed that culturing cells in 500 µM hydrocortisone increased the basal expression of CYP3A5 mRNA by 2-fold, masking the induction routinely observed using 10 µM BDP for 24 h. When A549 cells were subsequently cultured in media without hydrocortisone for 48 h, providing sufficient time for a “wash out” of the hydrocortisone, the basal expression of CYP3A5 mRNA was reduced, and ~2-fold induction of CYP3A5 mRNA occurred with the 10 µM BDP, 24 h treatment. Therefore, hydrocortisone was omitted from the SAEC growth media. Subsequent experiments in SAEC cells showed no change in CYP3A5 mRNA in response to glucocorticoid treatment (Figure 6), albeit removal of hydrocortisone from the media caused a slight decrease in the basal level of CYP3A5 mRNA expression, suggesting that GR plays a role in the regulation of CYP3A5. It is feasible that because cells had been exposed to such high concentrations of hydrocortisone during their isolation and expansion, that 10 µM of BDP was not sufficient to induce CYP3A5 mRNA, even after culturing the cells in the absence of hydrocortisone for multiple division cycles. Therefore, the concentration of BDP was increased to 50 µM and an additional treatment group using 150 µM [M1] was added. Again no increases in CYP3A5 mRNA was observed. Heat-inactivated and charcoal-stripped FBS were also utilized to remove potential interfering compounds from FBS, and still no change was observed.
To our knowledge, no one has observed a change in CYP3A mRNA expression in any primary human lung cell cultures. However, Cyp3a11, 3a13, and 3a16 mRNA and protein induction have been documented in mouse lung following dexamethasone treatment29. As such, additional studies using animal models and relevant samples from human patients need to be evaluated in order to conclusively confirm or reject the hypothesis that CYP3A genes are regulated in human subjects, particularly asthmatics, in response to glucocorticoid treatment since current in vitro models remain unexplainably limited in value for such studies.
In summary, the data presented herein demonstrate that, in A549 cells, glucocorticoid binding to the glucocorticoid receptor regulates the expression of CYP3A5. However, further research is needed to determine if changes in CYP3A5 expression occur in the human respiratory tissue similar to A549 cells, the precise mechanism by which this process occurs, and whether changes in the local metabolism of glucocorticoids by CYP3A5 ultimately impact glucocorticoid efficiency in asthma patients refractory to glucocorticoid treatment. Because we have not been able to evaluate this mechanism more thoroughly in primary lung cells, particularly from asthmatic subjects, the physiological and/or clinical relevance of the present study in steroid insensitive patients requires further investigation.
Author contributions
Roberts, Romero, Moore, Ward, Yost, and Reilly participated in research design. Roberts, Moore, and Romero conducted the experiments. Roberts, Moore, and Romero performed the data analysis. Roberts, Moore, Ward, Yost, and Reilly wrote or contributed to the writing of the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
The project described was supported by Grant Number R01HD060559 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health. J.K. Roberts was also supported by the Howard Hughes Medical Institute under their HHMI Med to Grad initiative [Grant 56006777].
Acknowledgments
We would like to thank Dr. Roger Gaedigk for the CYP3A5*1 probe and primer sequences.
Faculty Opinions recommendedReferences
- 1.
EPR-3. Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007.
J Allergy Clin Immunol.
2007; 120(5 Suppl): S94–138. PubMed Abstract
| Publisher Full Text
- 2.
Bateman ED, Hurd SS, Barnes PJ, et al.:
Global strategy for asthma management and prevention: GINA executive summary.
Eur Respir J.
2008; 31(1): 143–178. PubMed Abstract
| Publisher Full Text
- 3.
Williams SG, Schmidt DK, Redd SC, et al.:
Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program.
MMWR Recomm Rep.
2003; 52(RR-6): 1–8. PubMed Abstract
- 4.
Jusko WJ:
Corticosteroid pharmacodynamics: models for a broad array of receptor-mediated pharmacologic effects.
J Clin Pharmacol.
1990; 30(4): 303–310. PubMed Abstract
| Publisher Full Text
- 5.
Leung DY, Bloom JW:
Update on Glucocorticoid Action and Resistance.
J Allergy Clin Immunol.
2003; 111(1): 3–22. PubMed Abstract
| Publisher Full Text
- 6.
Mjaanes CM, Whelan GJ, Szefler SJ:
Corticosteroid therapy in Asthma: Predictors of Responsiveness.
Clin Chest Med.
2006; 27(1): 119–132. PubMed Abstract
| Publisher Full Text
- 7.
Wilcox JB, Avery GS:
Beclomethasone dipropionate corticosteroid inhaler: a preliminary report of its pharmacological properties and therapeutic efficacy in asthma.
Drugs.
1973; 6(2): 84–93. PubMed Abstract
| Publisher Full Text
- 8.
Jonsson G, Astrom A, Andersson P:
Budesonide is metabolized by cytochrome P450 3A (CYP3A) enzymes in human liver.
Drug Metab Dispos.
1995; 23(1): 137–142. PubMed Abstract
- 9.
Pearce RE, Leeder JS, Kearns GL:
Biotransformation of fluticasone: in vitro characterization.
Drug Metab Dispos.
2006; 34(6): 1035–1040. PubMed Abstract
| Publisher Full Text
- 10.
Moore CD, Roberts JK, Orton CR, et al.:
Metabolic pathways of inhaled glucocorticoids by the CYP3A enzymes.
Drug Metab Dispos.
2013; 41(2): 379–389. PubMed Abstract
| Publisher Full Text
- 11.
Roberts JK, Moore CD, Ward RM, et al.:
Metabolism of Beclomethasone Dipropionate by Cytochrome P450 3A Enzymes.
J Pharmacol Exp Ther.
2013; 345(2): 308–316. PubMed Abstract
| Publisher Full Text
- 12.
Leclerc J, Tournel G, Courcot-Ngoubo Ngangue E, et al.:
Profiling gene expression of whole cytochrome P450 superfamily in human bronchial and peripheral lung tissues: Differential expression in non-small cell lung cancers.
Biochimie.
2010; 92(3): 292–306. PubMed Abstract
| Publisher Full Text
- 13.
Westlind-Johnsson A, Malmebo S, Johansson A, et al.:
Comparative analysis of CYP3A expression in human liver suggests only a minor role for CYP3A5 in drug metabolism.
Drug Metab Dispos.
2003; 31(6): 755–761. PubMed Abstract
| Publisher Full Text
- 14.
Hukkanen J, Hakkola J, Anttila S, et al.:
Detection of mRNA encoding xenobiotic-metabolizing cytochrome P450s in human bronchoalveolar macrophages and peripheral blood lymphocytes.
Mol Carcinogen.
1997; 20(2): 224–230. PubMed Abstract
| Publisher Full Text
- 15.
Hukkanen J, Vaisanen T, Lassila A, et al.:
Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells.
J Pharmacol Exp Ther.
2003; 304(2): 745–752. PubMed Abstract
| Publisher Full Text
- 16.
Courcot E, Leclerc J, Lafitte JJ, et al.:
Xenobiotic metabolism and disposition in human lung cell models: comparison with in vivo expression profiles.
Drug Metab Dispos.
2012; 40(10): 1953–1965. PubMed Abstract
| Publisher Full Text
- 17.
Lacroix D, Sonnier M, Moncion A, et al.:
Expression of CYP3A in the human liver--evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth.
Eur J Biochem.
1997; 247(2): 625–634. PubMed Abstract
| Publisher Full Text
- 18.
Schuetz JD, Beach DL, Guzelian PS:
Selective expression of cytochrome P450 CYP3A mRNAs in embryonic and adult human liver.
Pharmacogenetics.
1994; 4(1): 11–20. PubMed Abstract
- 19.
Goodwin B, Redinbo MR, Kliewer SA:
Regulation of cyp3a gene transcription by the pregnane x receptor.
Annu Rev Pharmacol Toxicol.
2002; 42: 1–23. PubMed Abstract
| Publisher Full Text
- 20.
Kliewer SA, Goodwin B, Willson TM:
The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism.
Endocr Rev.
2002; 23(5): 687–702. PubMed Abstract
| Publisher Full Text
- 21.
Raunio H, Hakkola J, Pelkonen O:
Regulation of CYP3A genes in the human respiratory tract.
Chem Biol Interact.
2005; 151(2): 53–62. PubMed Abstract
| Publisher Full Text
- 22.
Dvorak Z, Pavek P:
Regulation of drug-metabolizing cytochrome P450 enzymes by glucocorticoids.
Drug Metab Rev.
2010; 42(4): 621–635. PubMed Abstract
| Publisher Full Text
- 23.
Urquhart BL, Tirona RG, Kim RB:
Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs.
J Clin Pharmacol.
2007; 47(5): 566–578. PubMed Abstract
| Publisher Full Text
- 24.
Schuetz JD, Schuetz EG, Thottassery JV, et al.:
Identification of a novel dexamethasone responsive enhancer in the human CYP3A5 gene and its activation in human and rat liver cells.
Mol Pharmacol.
1996; 49(1): 63–72. PubMed Abstract
- 25.
Duret C, Daujat-Chavanieu M, Pascussi JM, et al.:
Ketoconazole and miconazole are antagonists of the human glucocorticoid receptor: consequences on the expression and function of the constitutive androstane receptor and the pregnane X receptor.
Mol Pharmacol.
2006; 70(1): 329–339. PubMed Abstract
| Publisher Full Text
- 26.
Katome T, Obata T, Matsushima R, et al.:
Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions.
J Biol Chem.
2003; 278(30): 28312–28323. PubMed Abstract
| Publisher Full Text
- 27.
DeKeyser JG, Laurenzana EM, Peterson EC, et al.:
Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor.
Toxicol Sci.
2011; 120(2): 381–391. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Kuehl P, Zhang J, Lin Y, et al.:
Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression.
Nat Genet.
2001; 27(4): 383–391. PubMed Abstract
| Publisher Full Text
- 29.
Haag M, Fautrel A, Guillouzo A, et al.:
Expression of cytochromes P450 3A in mouse lung: effects of dexamethasone and pregnenolone 16alpha-carbonitrile.
Arch Toxicol.
2003; 77(3): 145–149. PubMed Abstract
Comments on this article Comments (0)