Introduction
The seminal and visionary work by Ohno in 1970 emphasized the pivotal role played by gene duplication in evolution. Gene duplication provides natural selection with the underlying mechanism to add functionality and adaptability to the genome by reusing pre-existing efficient and stable protein folds to catalyze novel reactions1. The fate of duplicated genes - unchanged functionality, pseudogenization, subfunctionalization or neofunctionalization - is the focus of intense research enabled by the advancements in sequencing technologies2,3. Differing opinions on function innovation have also been articulated4, and substantiated with real time evolution experiments5.
Gene duplication plays a key role in the evolution of virulence-associated genes6. Secreted lipases, one of the highly replicated genes in Candida albicans7, has been implicated in disease pathogenesis, both in humans8 and in plants9. A lipase/esterase (LipA) from X. oryzae (Xanthomonas orysae pv. oryzaeraises (Xoo)) that causes bacterial blight in rice and is conserved across the genus Xanthomonas, has been recently characterized10. LipA also has three homologs (LesA, LesB and LesC) in the Xylella fastidiosa (Xf) genome11.
Xf is a major source of concern for both economic12 and food security reasons13, being the causal agent for Pierce’s disease of grape (PD) and citrus variegated chlorosis (CVC) of citrus species14. The presence of three duplicated genes (LesA/B/C) closely related to LipA from X. oryzae raises certain intriguing questions. It is logical to assume that these genes serve different purposes, since it is unlikely that three genes with identical functions will be maintained in the genome. A deeper understanding of their respective roles in the phenotypic context is essential in order to develop novel strategies to counter their threat15–17. LesA and LesB have 93% identity - yet, LesA can hydrolyze tributyrin whereas LesB can not. In the current work, we aim to induce tributyrin hydrolysis in LesB using minimal mutations.
The desire to mimic and accelerate natural evolution has fueled interest in directed evolution experiments, which endow or enhance functionality in enzymes. There has been some pioneering work in applying de novo methods to obtain catalytic functions18–22. However, most methods start with a template protein having the desired activity, known active site residues and 3D structure23–25. Previously, we have established a computational method (CLASP) based on spatial and electrostatic properties for the detection of active sites26–29, and a methodology to quantify promiscuity in proteins30. We also explored the prospect of promiscuous active sites to serve as the starting point for directed evolution (DECAAF)31,32. DECAAF has been applied to the problem of identifying mutations in LesB based on the active site of LesA in order to endow LesB with tributyrin hydrolysis.
Since the structures of LesA/B are not known, and they share significant sequence homology with LipA (whose structure is known: PDBid:3H2G10), we used RaptorX to model the LesA/B structures. We first verified that the electrostatic profile of LesA and LesB are different. The LesA and LesB structures were then superimposed, and residues within a radii of 6 Å (MUT1:three residues) and 8 Å (MUT2:eight residues, including the three residues in MUT1) from the residues of the catalytic triad in LesA were compared to those in LesB. The differing residues were identified as the set of mutations which would induce tributyrin hydrolysis in LesB. It was observed that MUT1 and MUT2 residues are in two different contiguous stretches in the protein. As a validation step, we modeled the mutated sequences of LesB using RaptorX, and analyzed the differences in their electrostatic profiles. We created two mutants for LesB: LesBMUT1 with three mutations, and LesBMUT2 with eight mutations. The mutations in LesBMUT1 replicated the electrostatic congruence in the MUT1 residues, but not in the MUT2 residues. Consequently, we expected LesBMUT2 to have tributyrin hydrolysis, but not LesBMUT1.
We tested for the activity of LesB wild type and LesBMUT1/LesBMUT2 proteins using two assays. In one we use agar plates containing emulsified tributyrin, which upon hydrolysis of tributyrin to glycerol and butyric acid makes a clear zone visible. The other assay is a fluorescent quantitative assay in which 4-methylumbelliferyl butyrate (4-MUB) is used as a fluorescent substrate. The tributyrin hydrolysis activity of wild type LesB and LesBMUT1/LesBMUT2 with suggested mutations was tested in vitro using heterologous expression of these proteins in E. coli. We were able to confirm the tributyrin hydrolysis activity of LesBMUT2 in vitro using the assays mentioned above, while LesBMUT1 showed no activity.
Results and discussion
Dataset 1.LipA and LesA/B/C multiple sequence alignment and 4-MUB assay data.
This zip file contains: a multiple sequence alignment for LipA and LesA/B/C (ALN format), and a CSV, which contains raw values for 4-MUB assay presenting relative fluorescent units (RFU). EV, empty vector; PBS, phosphate buffered saline.The sequence alignment for LipA10, LesA (PD1703), LesB (PD1702) and LesC (P D1211) is shown in Figure 1a (gene names from http://www.ncbi.nlm.nih.gov/gene/). The phylogenetic tree and the pairwise sequence identity and similarity (Supplementary Table 1) suggests that LipA is more closely related to LesA/B than to LesC (Figure 1b). For example, LesA and LesB have 90% identity (347 out of 387 residues are identical) and 93% similarity (361 out of 387 residues are similar). In lieu of these differences in LesB, it does not have the capability to hydrolyze tributyrin that LesA does.
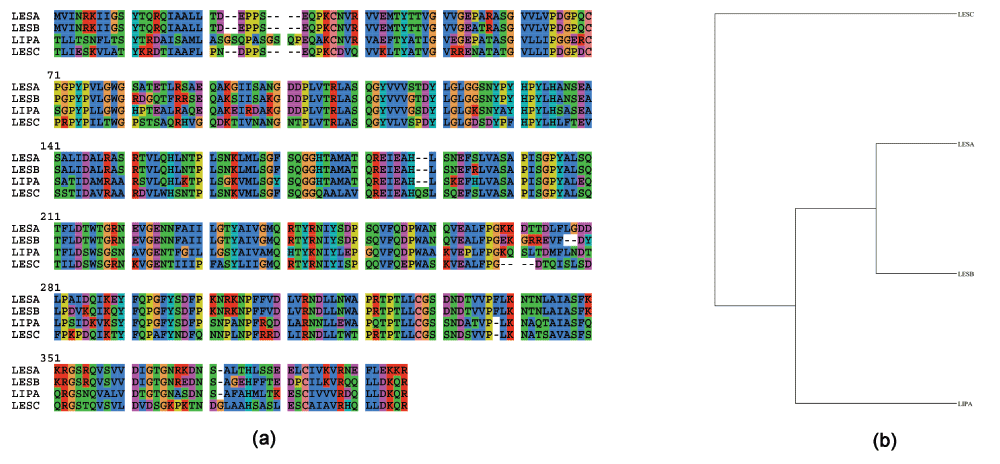
Figure 1. Multiple sequence alignment done using ClustalW, and phylogenetic trees generated using PhyML for LipA and LesA/B/C.
(a) Sequence alignment. (b) Cladogram generated from (a).
The structures for LesA/B/C have not been solved. However, since there is significant sequence homology of these proteins with LipA, we modeled the structures of these proteins using RaptorX33. RaptorX automatically chooses the best template (LesA - PDBid:3H2G, LesB and LesC - PDBid:3H2I)10. The structural superimposition of these proteins done using MUSTANG34 is shown in Figure 2a.
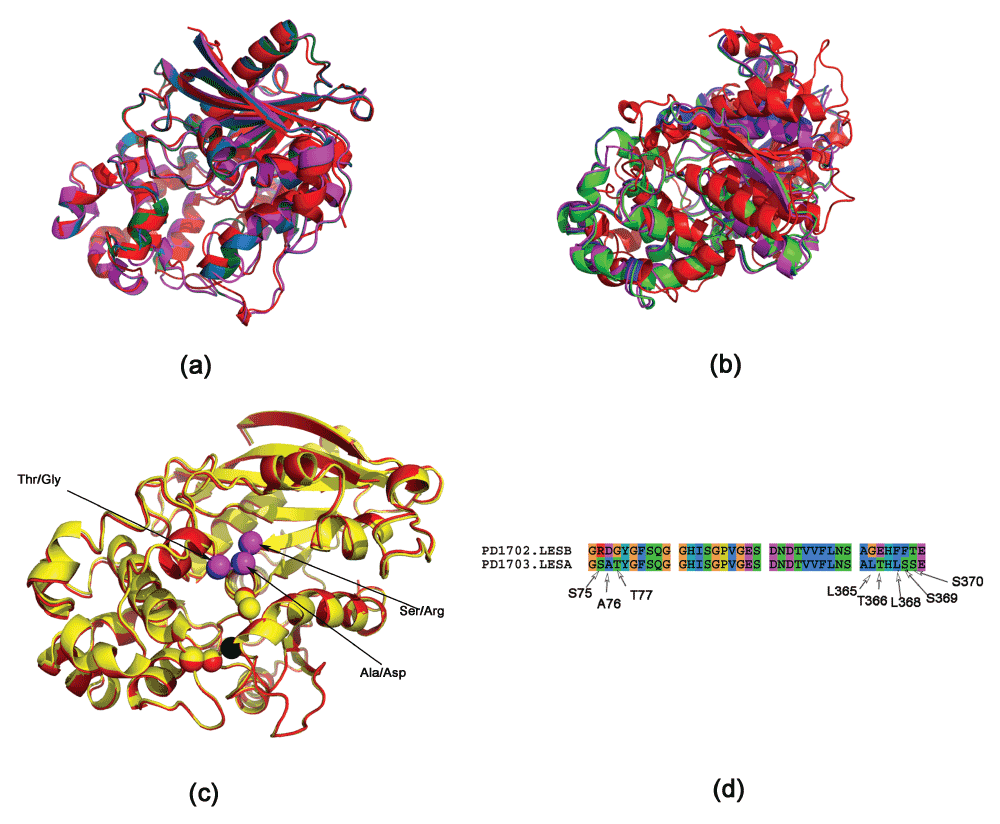
Figure 2. Superimposing LipA and LesA/B/C proteins.
(a) MUSTANG generated superimposition (LipA - red, LesA - green, LesB - blue, LesC - magenta). (b) STEEP generated superimposition, obtained by superimposing three atoms from the catalytic triad in the active site. It can be seen that MUSTANG generates a better overall superimposition, since STEEP tries to get a better superimposition of the atoms, rather than achieve a global superimposition32. (c) Superimposition of LesA and LesB (red and yellow, respectively), obtained by superimposing the catalytic triad (Asp336, Ser176 and His377 in LipA). The Asp overlaps completely (at the origin of the coordinate system) and is shown in black. Some of the different residues are shown in magenta and blue for LesA and LesB respectively. (d) Sequence alignment of active site residues (residues within a radii of 7 Å from the residues of the catalytic triad in LesA). Note that these residues are not in a one contiguous stretch.
The active sites of these proteins are conserved and have a serine catalytic triad (Asp336, Ser176 and His377 in LipA). Previously, we have proposed a method to suggest mutations in a protein in order to endow it with a specific catalytic function based on structural and electrostatic homology of residues in the active site to a known protein with the desired function31. We chose the Cβ atoms of the residues as the representative atom for each residue for an uniform comparison (only glycine lacks a Cβ atom). We have shown that although the reactive groups are different for amino acids, this difference is encapsulated in the backbone Cβ atoms35. Table 1 shows the spatial and electrostatic potential difference (EPD) congruence in the catalytic triad in these proteins. We applied transformations to align the catalytic triad (Figure 2b)31.
Table 1. Potential and spatial congruence of the active site residues in LesA/B and the mutant LesB’s (LesBMUT1 and LesBMUT2).
It can be seen that the serine catalytic triad is congruent in all proteins. Further, LesA and LesB have a different electrostatic profile when we consider another residue (Ser75/Arg75 respectively) - see pair ‘bd’ for instance. While the EPD in this pair is moderately positive in LesA (+38.5 EPD units), this is seen to change sign in LesB (-63.1 EPD units). The mutated structures (LesBMUT1 and LesBMUT2) modulates this EPD back to moderately positive again (+19 and +12 units in LesBMUT1 and LesBMUT2, respectively). D = Pairwise distance in Å. PD = Pairwise potential difference. See Methods section for units of potential.
| PDB | Active site atoms
(a,b,c,d) | | ab | ac | ad | bc | bd | cd |
|---|
| LesA | ASP325CB,SER165CB,
HIS367CB,SER75CB, | D
PD | 9.9
-61.9 | 4.9
-113.8 | 17.6
-23.5 | 7.0
-51.9 | 8.0
38.5 | 14.2
90.4 |
| LesB | ASP323CB,SER165CB,
HIS365CB,ARG75CB, | D
PD | 9.8
-48.8 | 5.1
-126.8 | 17.5
-111.9 | 6.8
-78.0 | 8.1
-63.1 | 14.0
14.9 |
| LesBMUT1 | ASP322CB,SER164CB,
HIS364CB,SER74CB, | D
PD | 9.8
-90.3 | 4.9
-118.2 | 17.5
-71.0 | 6.8
-27.9 | 8.1
19.3 | 14.0
47.2 |
| LesBMUT2 | ASP323CB,SER165CB,
HIS365CB,SER75CB, | D
PD | 9.9
-69.4 | 5.1
-114.1 | 17.7
-57.0 | 6.9
-44.7 | 8.2
12.4 | 14.2
57.1 |
This multiple superimposition of the proteins provided a single frame of reference for comparing the proteins LesA and LesB (Figure 2c). After the superimposition, we took residues within a radii of 7 Å from the residues of the catalytic triad in LesA. Now, for each of these residues we found the closest residue in LesB - noting that they are now superimposed. The different residues, that form the set which are to be mutated, are shown in Table 2 and Figure 2d. It can be seen that these residues lie within two contiguous stretches: a) R75, D76 and G77 (MUT1) which are about 6 Å away from the active site residues and b) G363, E364, F366, F367 and T368 (MUT2) that are about 8 Å away from the active site residues. We created a LesB mutant by mutating resides in (a) (LesBMUT1), and another (LesBMUT2) by mutating residues in both sets.
Table 2. Mutations required to mimic the LesA active site in the LesB protein.
Two sets of mutations were studies here - LesBMUT1 and LesBMUT2. The LesBMUT1 mutations are within 6 Å of the active site residues, and located within a contiguous stretch. We further added the LesBMUT1 mutations to another set of residues that differ (8 Å away from the active site residues) to obtain LesBMUT2 mutations.
| LesB | LesA | LesBMUT1 | LesBMUT2 |
|---|
| R75 | S75 | S75 | S75 |
| D76 | A76 | A76 | A76 |
| G77 | T77 | T77 | T77 |
| G363 | L365 | - | L363 |
| E364 | T366 | - | T364 |
| F366 | L368 | - | L366 |
| F367 | S369 | - | S367 |
| T368 | S370 | - | S378 |
As a validation step, we modeled the structures of the mutated sequences in order to compare the change in the electrostatic profile. We first consider the catalytic triad and one of the mutated amino acid (Arg75 to Ser75 in LesB) in the LesBMUT1/LesBMUT2 structures (Table 1). It can be seen that the mutated active site is spatially similar to wild type. The same holds true for all structures discussed henceforth. The major change in the electrostatic profile can be seen for the pair ‘bd’ (Ser165CB/Arg75CB in LesB versus Ser165CB/Ser75CB in LesBMUT1/LesBMUT12). While the EPD in this pair is moderately positive in LesA (+38.5 EPD units), this is seen to change sign in LesB (-63.1 EPD units). The mutated structures (LesBMUT1/LesBMUT2) modulate this EPD back to moderately positive again (+12.4 EPD units).
While it may appear from Table 1 that the three mutations (MUT1) in LesBMUT1 might suffice, it is necessary to compute the changes in the MUT2 residues. We chose one residue from the active site (Asp323) and three residues from the MUT2 set (E364, F366 and F367). Since Gly does not have a Cβ atom, it could not be used for comparison. There are certain differences in the electrostatic profile between LesBMUT1 and LesBMUT2. While the EPD in ‘bd’ is moderately positive in LesA (+8.2 EPD units), this changes sign in LesBMUT1 (-72.3 EPD units) (Table 3). However, this EPD modulates back to moderately positive again in LesBMUT2. Thus, it might be necessary to introduce all eight mutations (MUT1 + MUT2) in order to introduce tributyrin hydrolysis in LesB. This verification step is unique to our methodology, and infuses confidence in the chances of success with the in vivo mutations. We now made two mutants of LesB - LesBMUT1 having three mutations, and LesBMUT2 having eight mutations.
Table 3. Potential and spatial congruence of the active site residues in LesA and the two LesB variants (LesBMUT1 and LesBMUT2).
We chose one residue from the active site (Asp323) and three residues from the MUT2 set (E364, F366 and F367). Since Gly does not have a Cβ atom, it could not be used for comparison. We observed certain differences in the electrostatic profile between LesBMUT1 and LesBMUT2 - see pair ‘bd’ for instance. While the EPD in this pair is moderately positive in LesA (+8.2 EPD units), this is seen to change sign in LesBMUT1 (-72.3 EPD units). This EPD modulates back to moderately positive again in LesBMUT2. Thus, it might be necessary to introduce all eight mutations (MUT1 + MUT2) in order to introduce tributryn catalysis in LesB. D = Pairwise distance in Å. PD = Pairwise potential difference. See Methods section for units of potential.
| PDB | Active site atoms
(a,b,c,d) | | ab | ac | ad | bc | bd | cd |
|---|
| LESA | ASP325CB,THR366CB,
LEU368CB,SER369CB, | D
PD | 5.2
-20.8 | 9.4
-38.6 | 8.4
-12.6 | 7.2
-17.8 | 5.2
8.2 | 5.4
26.0 |
| LESBMUT1 | ASP322CB,GLU363CB,
PHE365CB,PHE366CB, | D
PD | 4.8
-8.6 | 9.3
-118.2 | 8.4
-80.9 | 7.5
-109.6 | 5.5
-72.3 | 5.5
37.3 |
| LESBMUT2 | ASP323CB,THR364CB,
LEU366CB,SER367CB, | D
PD | 4.9
-13.4 | 9.5
-44.1 | 8.5
5.4 | 7.5
-30.7 | 5.4
18.9 | 5.4
49.6 |
To test the activity of LesBMUT1/LesBMUT2 in vitro, we expressed LesB and LesBMUT1/LesBMUT2 in E. coli using an expression vector to obtain high quantities of heterologous protein. The E. coli strain expressing LesBMUT2 streaked on tributyrin agar plates showed significant tributyrin hydrolysis activity compared to wild type LesB. The activity was visualized by a tributyrin hydrolysis zone on the agar plate containing emulsified tributyrin fat (Figure 3a). However, the LesBMUT1 showed no tributyrin hydrolysis under the same conditions.
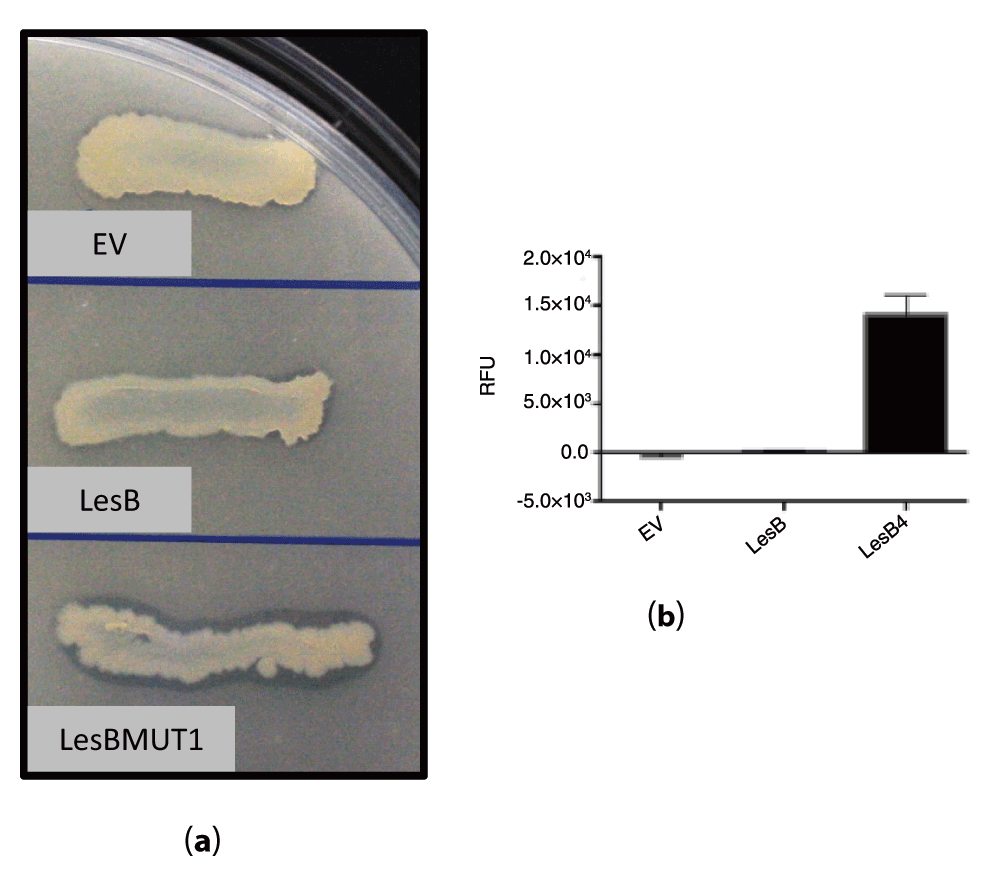
Figure 3. Assays to detect tributyrin hydrolysis activity for LesB, LesBMUT2 and EV (empty vector).
(a) Tributyrin agar plate assay. Tributyrin hydrolysis zone (clearing zone) is visible for E. coli strains expressing LesBMUT2 and not for LesB or empty vector (EV) strains. (b) A4-Methylumbelliferyl butyrate (4-MUB) fluorescent assay detecting tributyrin hydrolysis activity of LesB, LesBMUT2 and EV (empty vector) proteins expressed in E. coli.
Using 4-MUB assay we were able to detect tributyrin hydrolysis activity and confirm what we observed in tributyrin agar plate. Crude protein extracted from LesBMUT2 expressing E. coli showed a significant increase in tributyrin hydrolysis activity as depicted in Figure 3b.
Asn228 in LipA is an interesting residue in the context of tributyrin hydrolysis. This residue is involved in the binding of glycoside detergent β-octyl glucoside (BOG) in LipA (PDBid:3H2K) in X. oryzae pv oryzae. Interestingly, while the mutant N228W mutant (PDBid:3H2I) is still able to hydrolyze tributyrin, it showed virulence deficiency similar to that of the LipA-deficient strain BXO200136. Corroborating the redundancy of Asn228 in tributyrin hydrolysis, we note that this residue falls outside the 8 Å radius chosen for LesBMUT2 mutations, which were able to induce tributyrin hydrolysis.
Materials and methods
Heterologous protein expression in E. coli
In order to test for tributyrin hydrolysis activity, the new open reading frame with suggested mutations as well as wild type LesB was codon optimized for expression in E. coli and were chemically synthesized (DNA2.0, Menlo Park, CA). The resulting coding sequences were cloned into pJ401: T5 expression vector from DNA2.0 to obtain high amounts of recombinant LesB4 and LesB.
Cell culture and protein extraction
E. coli strains expressing LesB wild type and LesB4 as well as empty vector control (EV) were inoculated into liquid cultures and grown overnight at 37°C with constant shaking at 200 rpm. The overnight cultures were added to a larger flask with fresh LB media and grown under the same conditions until they reached OD of 0.5–0.8, at the point which 0.3 mM of IPTG (Sigma Aldrich) was added to each culture to induce the promoter. Induced cultures were incubated at room temperature with constant shaking at 200 RPM overnight. The following day cultures were spun down at 5000g (Sigma 3K10) to pellet the cells and resulting pellet was suspended in sterile PBS. Next, the obtained cells in PBS were lysed using a microfluidizer (Microfluidics M-110L) machine. The extracted protein was quantified and used for the 4-MUB assay.
4-Methylumbelliferyl butyrate (4-MUB) assay
The quantitative detection of LesA enzyme was carried out using MUB Assay based on Vaneechoutte et al.37. For each samples 3 technical replicates were used. Briefly, A stock solution of the substrate, made by dissolving 10 mg of 4-methylumbelliferyl butyrate (4-MUB) (Sigma Chemical Co., St. Louis, MO) in 1 mL of dimethyl sulfoxide (DMSO) and 10 μL of Triton X-100 to obtain 40 mM stock solution which was further diluted to 5 mM using 0.1 M citrate buffer (pH 5.0). For each reaction 80 μL of substrate was added to 20 μL of each sample immediately before the fluorescent intensity was read in a fluorometer Plate Reader SpectraMax M2 (Molecular Devices) at 365 nm excitation and 455 nm emission at 30°C for 30 minutes. Fluorescence values measured in 4-MUB assay were used to calculate mean and standard deviations. After subtracting the background (PBS) values were plotted using Prism version 6.0c.
In silico methods.
The current work provides in vivo validation of previously described in silico methods26,31,32. The underlying theoretical foundation for our methods is the non-triviality of the spatial and electrostatic congruence in cognate pairs seen across various structures of the same catalytic function. We identify spatially equivalent residues that have differing electrostatic properties, based on the logic that functional divergence in the protein family arises from these residues.
In order to superimpose the two scaffolds - LesA=(Asp336, Ser176 and His377) and LesB=(Asp323, Ser165 and His365) - we applied linear and rotational transformations for all atoms in LesA and LesB such that the Cβ atoms of all residues lay on the same plane (Z=0), and were at the center, and and
lie on the Y axis. This superimposition was outputted as a Pymol formatted file.
We aligned the residues from LesA which are within 7 Å radius from the active site residues (Asp336, Ser176 and His377). The choice of the radial distance that encompasses interacting residues is critical, since a small radius will not include enough residues, and a large one will include irrelevant ones. Next, for LesB we identified residues that were in the vicinity of each of the residues in LesA, choosing the closest residue as the alignment for the position p. This is possible since we have a consolidated spatial reference frame for both the proteins. RaptorX was then used to predict the structure of the mutated LesB33.
Adaptive Poisson-Boltzmann Solver (APBS) and PDB2PQR packages were used to calculate the potential difference between the reactive atoms of the corresponding proteins38,39. The APBS parameters and electrostatic potential units were set as described previously in26.
All protein structures were rendered by PyMol (http://www.pymol.org/). The alignment and cladograms images were created using Seaview40. PHYML was used to generate phylogenetic trees from these alignments, which searches for a tree with the highest probability or likelihood that, given a proposed model of evolution and the hypothesized history, would give rise to the observed data set (method of maximum likelihood)41.
Data availability
F1000Research: Dataset 1. LipA and LesA/B/C multiple sequence alignment and 4-MUB assay data, 10.5256/f1000research.5147.d3495442
Author contributions
HG performed the experiments. SC wrote the computer programs. All authors analyzed the data, and contributed equally to the writing and subsequent refinement of the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant. BA acknowledges financial support from the Science Institute of the University of Iceland.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Conant GC, Wolfe KH:
Turning a hobby into a job: how duplicated genes find new functions.
Nat Rev Genet.
2008; 9(12): 938–950. PubMed Abstract
| Publisher Full Text
- 2.
Lynch M, Conery JS:
The evolutionary fate and consequences of duplicate genes.
Science.
2000; 290(5494): 1151–1155. PubMed Abstract
| Publisher Full Text
- 3.
Zhang J:
Evolution by gene duplication: an update.
Trends Ecol Evol.
2003; 18(6): 292–298. Publisher Full Text
- 4.
Bergthorsson U, Andersson DI, Roth JR:
Ohno’s dilemma: evolution of new genes under continuous selection.
Proc Natl Acad Sci U S A.
2007; 104(43): 17004–17009. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Nasvall J, Sun L, Roth JR, et al.:
Real-time evolution of new genes by innovation, amplification, and divergence.
Science.
2012; 338(6105): 384–387. PubMed Abstract
| Publisher Full Text
- 6.
Moran GP, Coleman DC, Sullivan DJ:
Comparative genomics and the evolution of pathogenicity in human pathogenic fungi.
Eukaryotic Cell.
2011; 10(1): 34–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Hube B, Stehr F, Bossenz M, et al.:
Secreted lipases of Candida albicans: cloning, characterisation and expression analysis of a new gene family with at least ten members.
Arch Microbiol.
2000; 174(5): 362–374. PubMed Abstract
| Publisher Full Text
- 8.
Gacser A, Trofa D, Schafer W, et al.:
Targeted gene deletion in Candida parapsilosis demonstrates the role of secreted lipase in virulence.
J Clin Invest.
2007; 117(10): 3049–3058. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Voigt CA, Schafer W, Salomon S:
A secreted lipase of Fusarium graminearum is a virulence factor required for infection of cereals.
Plant J.
2005; 42(3): 364–375. PubMed Abstract
| Publisher Full Text
- 10.
Aparna G, Chatterjee A, Sonti RV, et al.:
A cell wall-degrading esterase of Xanthomonas oryzae requires a unique substrate recognition module for pathogenesis on rice.
Plant Cell.
2009; 21(6): 1860–1873. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Simpson AJ, Reinach FC, Arruda P, et al.:
The genome sequence of the plant pathogen Xylella fastidiosa. The Xylella fastidiosa Consortium of the Organization for Nucleotide Sequencing and Analysis.
Nature.
2000; 406(6792): 151–159. PubMed Abstract
| Publisher Full Text
- 12.
Alston JM, Fuller KB, Kaplan JD, et al.:
Assessing the returns to R&D on perennial crops: the costs and benefits of pierce’s disease research in the california winegrape industry.
Aust J Agric Resour Econ.
2014. Publisher Full Text
- 13.
Strange RN, Scott PR:
Plant disease: a threat to global food security.
Annu Rev Phytopathol.
2005; 43: 83–116. PubMed Abstract
| Publisher Full Text
- 14.
Hopkins D, Purcell A:
Xylella fastidiosa: cause of Pierce’s disease of grapevine and other emergent diseases.
Plant Disease.
2002; 86(10): 1056–1066. Publisher Full Text
- 15.
Gray D, Li Z, Hopkins D, et al.:
Transgenic grapevines resistant to Pierce’s disease.
HortScience.
2005; 40(4): 1104–1105. Reference Source
- 16.
Agueero CB, Uratsu SL, Greve C, et al.:
Evaluation of tolerance to Pierce’s disease and botrytis in transgenic plants of Vitis vinifera l. expressing the pear pgip gene.
Mol Plant Pathol.
2005; 6(1): 43–51. PubMed Abstract
| Publisher Full Text
- 17.
Dandekar AM, Gouran H, Ibanez AM, et al.:
An engineered innate immune defense protects grapevines from Pierce disease.
Proc Natl Acad Sci U S A.
2012; 109(10): 3721–3725. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Bolon DN, Mayo SL:
Enzyme-like proteins by computational design.
Proc Natl Acad Sci U S A.
2001; 98(25): 14274–14279. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 19.
Jiang L, Althoff EA, Clemente FR, et al.:
De novo computational design of retro-aldol enzymes.
Science.
2008; 319(5868): 1387–1391. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Faiella M, Andreozzi C, de Rosales RT, et al.:
An artificial di-iron oxo-protein with phenol oxidase activity.
Nat Chem Biol.
2009; 5(12): 882–884. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Siegel JB, Zanghellini A, Lovick HM, et al.:
Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction.
Science.
2010; 329(5989): 309–313. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Rothlisberger D, Khersonsky O, Wollacott AM, et al.:
Kemp elimination catalysts by computational enzyme design.
Nature.
2008; 453(7192): 190–195. PubMed Abstract
| Publisher Full Text
- 23.
Reetz MT, Carballeira JD:
Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes.
Nat Protoc.
2007; 2(4): 891–903. PubMed Abstract
| Publisher Full Text
- 24.
Climie S, Ruiz-Perez L, Gonzalez-Pacanowska D, et al.:
Saturation site-directed mutagenesis of thymidylate synthase.
J Biol Chem.
1990; 265(31): 18776–18779. PubMed Abstract
- 25.
Reetz MT, Carballeira JD, Peyralans J, et al.:
Expanding the substrate scope of enzymes: combining mutations obtained by CASTing.
Chemistry.
2006; 12(23): 6031–6038. PubMed Abstract
| Publisher Full Text
- 26.
Chakraborty S, Minda R, Salaye L, et al.:
Active site detection by spatial conformity and electrostatic analysis--unravelling a proteolytic function in shrimp alkaline phosphatase.
PLoS One.
2011; 6(12): e28470. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Chakraborty S, Ásgeirsson B, Minda R, et al.:
Inhibition of a cold-active alkaline phosphatase by imipenem revealed by in silico modeling of metallo-β-lactamase active sites.
FEBS Lett.
2012; 586(20): 3710–3715. PubMed Abstract
| Publisher Full Text
- 28.
Rendon-Ramirez A, Shukla M, Oda M, et al.:
A computational module assembled from different protease family motifs identifies PI PLC from Bacillus cereus as a putative prolyl peptidase with a serine protease scaffold.
PLoS One.
2013; 8(8): e70923. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Chakraborty S, Rendon-Ramirez A, Ásgeirsson B, et al.:
Dipeptidyl peptidase-IV inhibitors used in type-2 diabetes inhibit a phospholipase c: a case of promiscuous scaffolds in proteins [v1; ref status: approved 1, approved with reservations 1, http://f1000r.es/2hw].
F1000Research.
2013; 2: 286. Reference Source
- 30.
Chakraborty S, Rao BJ:
A measure of the promiscuity of proteins and characteristics of residues in the vicinity of the catalytic site that regulate promiscuity.
PLoS One.
2012; 7(2): e32011. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Chakraborty S:
An automated flow for directed evolution based on detection of promiscuous scaffolds using spatial and electrostatic properties of catalytic residues.
PLoS One.
2012; 7(7): e40408. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Chakraborty S, Rao BJ, Baker N, et al.:
Structural phylogeny by profile extraction and multiple superimposition using electrostatic congruence as a discriminator.
Intrinsically Disordered Proteins.
2013; 1: e25463. Publisher Full Text
- 33.
Peng J, Xu J:
RaptorX: exploiting structure information for protein alignment by statistical inference.
Proteins.
2011; 79(Suppl 10): 161–171. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Konagurthu AS, Whisstock JC, Stuckey PJ, et al.:
MUSTANG: a multiple structural alignment algorithm.
Proteins.
2006; 64(3): 559–574. PubMed Abstract
| Publisher Full Text
- 35.
Chakraborty S, Rao BJ, Ásgeirsson B, et al.:
The electrostatic profile of consecutive Cβ atoms applied to protein structure quality assessment [v2; ref status: awaiting peer review, http://f1000r.es/2cf].
F1000Research.
2013; 2: 243. Publisher Full Text
- 36.
Rajeshwari R, Jha G, Sonti RV:
Role of an in planta-expressed xylanase of Xanthomonas oryzae pv. oryzae in promoting virulence on rice.
Mol Plant Microbe Interact.
2005; 18(8): 830–837. PubMed Abstract
| Publisher Full Text
- 37.
Vaneechoutte M, Verschraegen G, Claeys G, et al.:
Rapid identification of Branhamella catarrhalis with 4-methylumbelliferyl butyrate.
J Clin Microbiol.
1988; 26(6): 1227–1228. PubMed Abstract
| Free Full Text
- 38.
Baker NA, Sept D, Joseph S, et al.:
Electrostatics of nanosystems: application to microtubules and the ribosome.
Proc Natl Acad Sci U S A.
2001; 98(18): 10037–10041. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Dolinsky TJ, Nielsen JE, McCammon JA, et al.:
PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations.
Nucleic Acids Res.
2004; 32(Web Server issue): W665–667. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 40.
Gouy M, Guindon S, Gascuel O:
SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building.
Mol Biol Evol.
2010; 27(2): 221–224. PubMed Abstract
| Publisher Full Text
- 41.
Guindon S, Lethiec F, Duroux P, et al.:
PHYML Online--a web server for fast maximum likelihood-based phylogenetic inference.
Nucleic Acids Res.
2005; 33(Web Server issue): W557–559. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 42.
Gouran H, Chakraborty S, Rao BJ, et al.:
LipA and LesA/B/C multiple sequence alignment and 4-MUB assay data.
F1000Research.
2014. Data Source
Comments on this article Comments (0)