Introduction
The rapid development of crystallization techniques has resulted in a deluge of proteins with known structures1. Most of the proteins are annotated using sequence alignment methods by a ‘guilt by association’ logic based on the sequence-to-structure-to-function paradigm2. However, sequence alignment methods are not applicable in cases where similar functional groups are identically positioned in the active site of proteins with no sequence homology. The classic example of this phenomenon, known as convergent evolution3,4, is the major families of serine proteases (chymotrypsin and subtilisin), where the active site is structurally and functionally identical, though there is no global sequence or structural homology5. According to some studies, about 42% of entries annotated as ‘unknown functions’ are true examples of proteins of unknown function6.
Structure-based methods have evolved to detect such convergently evolved proteins7,8. The conservation of structural properties is the primary driving logic behind many of these identification methods, reviewed in detail previously9. There are essentially two categories of programs that find binding sites in proteins (binding sites are typically closely related to protein function). The first one requires a predefined set of amino acids (motifs) of a known enzymatic function to search for the same within the protein under investigation8,10–14. The second category automatically detects similarity in the side chain patterns to classify protein functionality7,15–18. We have demonstrated through several detailed examples, using a method (CLASP19) which falls in the first category, that such structural conservation necessitates the conservation of electrostatic properties in proteins with the same functionality19–22.
A challenge emerging in these methods relates to the large fold space of known proteins, although the rate of increase of this space is gradually being saturated23. Efficient parallelization has allowed the ProBiS algorithm to compare a protein query against the PDB in minutes15. To date, the identification of motifs is a task executed on the fly and applied sequentially13,14. Thus, running multiple queries involves several invocations of the same program. Our aim is to amortize the processing times by a one-time precompilation of all possible motifs, pruned using rational distance constraints, which can be leveraged for future queries.
A simplistic approach to obtain motifs is to enumerate all possible combinations from the sequence. Motifs that span across distances rarely seen in active sites can be pruned out using structural information. In the present work, we propose a method to precompile all possible motifs comprising of a set (n=4 in this case) of predefined amino acid residues from a protein structure that occur within a specified distance (R) of each other (PREMONITION - Preprocessing motifs in protein structures for search acceleration). We have estimated R from the known active site residues of ~500 proteins annotated in the CSA database (http://www.ebi.ac.uk/thornton-srv/databases/CSA/)24. PREMONITION rolls a sphere of radius R along the protein fold centered at the Cα atom of each residue, and all motifs are extracted within this sphere. The maximum number of residues that occurs within R Å of any residue in protein dataset is also computed. This sets an upper bound for the polynomial complexity of the PREMONITION algorithm, and run times for the precompilation are reasonable. After such a precompilation step, the computational time required for querying a protein structure with multiple motifs is reduced considerably.
Previously, we had proposed a computational method (PROMISE) to estimate the promiscuity of proteins with known active site residues and 3D structure using the CSA database25. It took less than a minute to query one protein with all 500 motifs using PREMONITION, a process that took almost a day when done sequentially. Such speed up enables querying a much larger set of proteins using a comprehensive set of ligands, as is required in drug screening procedures.
Materials and methods
Algorithm 1 details the steps in creating the PREMONITION database for a given protein. We enumerate the steps using a concrete example for trypsin (PDBid:1A0J). Given a radius of interaction SOAS, we first compute the set residues within SOAS for each residue Residuei. For example, let us assume we are processing residue D102. Equation 1 gives the set of residues that have at least one atom within SOAS=10 Å from the Cα of D102.
= [D102, W237, M90, V227, M180, G38, R62, S37, D194...] (1)
We now take combinations of n=4 from this set, which does not necessarily include D102 (Equation 2).
= [(W237, M90, D102, V227), (W237, M90, D194, M180), (G38, R62, S37, D102)...] (2)
After sorting the each combination based on the single letter amino acid code we obtain a set of n tuples (Equation 3).
= [DMVW = (102, 90, 227, 237), DDQV = (194, 90, 180, 237), DGRS = (102, 38, 62, 37)...] (3)
Now, we add (102,90,227,237) to the global tableofmatches for the key ‘DMVW’. Thus, as we process every residue in the protein, we merge all occurrences of ‘DMVW’ (Equation 4).
= [(194.104.53.141), (102.90.227.237), (194.104.52.51), (194.104.53.51)...] (4)
Extracting all motifs of ‘DMVW’ now consists of the trivial task of reading this set from the file on disk.
Adaptive Poisson-Boltzmann Solver (APBS) and PDB2PQR packages were used to calculate the potential difference between the reactive atoms of the corresponding proteins26,27. The APBS parameters and electrostatic potential units were set as described previously in19. Protein structures were rendered by PyMol (http://www.pymol.org/). The proteins were superimposed based on the matching motifs using DECAAF28.
Algorithm 1. Premonition()
Input: P1 : Reference protein
Input: n: number of residues in the motif
Input: SOAS: Radius of sphere centered around each residue
begin
/* Table mapping string of amino acids of length n to list of indices */
tableofmatches = ∅ ;
ϕca = Cα atoms of all residues ;
foreach CAi in ϕca do
ϕNearestResidues = FindResiduesWithinDist(CAi, SOAS);
ϕCombinations = GetCombinationsof_n(ϕNearestResidues, n);
ϕSortedStrings = SortBasedOnAminoAcidName(ϕSortedCombinations);
InsertInTable(ϕSortedStrings,tableofmatches);
end
/* Output in file */
foreach string in tableofmatches do
= GetMotifsforEachString(tableofmatches);
PrintListofMatches();
end
end
Results and discussion
Estimating the maximum radius for computing interacting residues
First, we estimated the minimal radius of a sphere that encompasses active sites found in proteins. CSA provides catalytic residue annotation for enzymes in the PDB and is available online24. The database consists of an original hand-annotated set extracted from the primary literature and a homologous set inferred by PSI-BLAST2. We chose ~500 proteins from the CSA database that are annotated from the literature (SI list.doc). We computed the size of the active site (SOAS) for these known active sites by finding the minimum radius centered around one residue that encompassed the other residues. Table 1 shows the pairwise distance for the four residues that comprise the active site in trypsin (PDBid:1A0J) - Asp102, Ser195, His57 and Ala56. It can be seen that a sphere of radius 6.9 Å centered around His57 (c) would include all other residues. Other radii required to encompass all other residues of the motif and centered at a different residue is larger than this value (Asp102 = 7.8 Å, Ser195 = 9 Å and Ala56 = 9 Å). Thus, the SOAS for this protein is 6.9 Å. Figure 1a shows the frequency distribution of the SOAS for the set of 500 proteins chosen (mean = 7.3 Å, standard deviation = 1.8 Å, min = 3.5 Å and max = 13 Å). 90% of proteins have an SOAS below 10 Å.
Table 1. Pairwise distance between the active site residues in trypsin (PDBid:1A0J).
The motif consists of Asp102/OD1(a), Ser195/OG(b), His57/ND1(c) and Ala56/N(d). A sphere of radius 6.9 Å centered around His57 (c) would include all other active site residues. This is the minimal distance - the radius of any sphere needed to include all residues is more than this value.
| Atom1 | Atom2 | Distance(Å) |
|---|
| a | b | 7.8 |
| a | c | 5.6 |
| a | d | 2.9 |
| b | c | 3.3 |
| b | d | 9.0 |
| c | d | 6.9 |
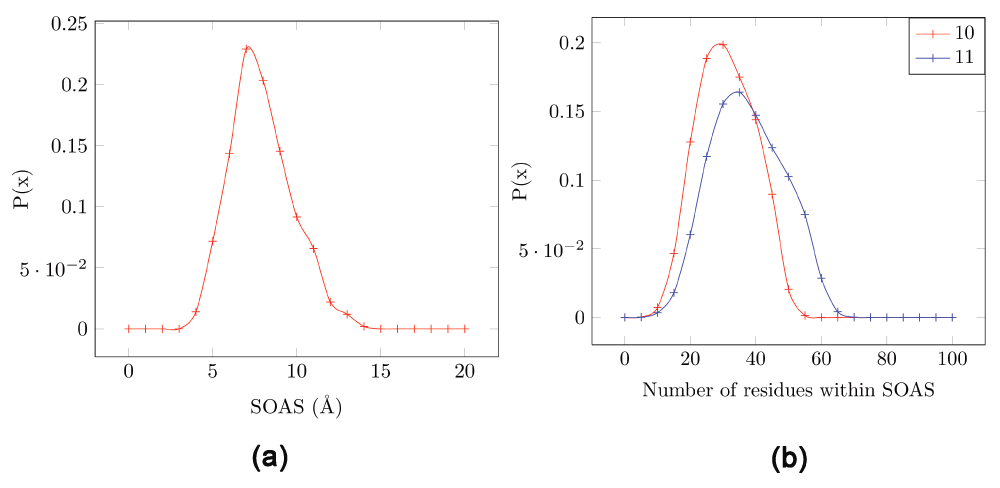
Figure 1. Estimating the radius of the sphere enclosing the active site in proteins, and the number of residues in the sphere.
Data is extracted from ~500 proteins from the CSA database which are annotated from literature. (a) The size of the active site (SOAS) computed using the minimum radius centered around one residue that encompasses the other residues. (b) Number of residues enclosed by the SOAS (Red=10 Å, blue=11 Å). A residue Rj is considered to be within the SOAS of another residue Ri if any atom of Rj falls within a sphere of radius SOAS centered around the Cα of Ri.
Number of residues within a sphere of radius 10 Å
Next, we estimated the number of residues that fall within the SOAS radius in different proteins. PREMONITION takes combinations of 4 residues from each of this set (size N), and is thus polynomial in complexity (O(N) = N4). The value of upper bound on N needs to be known to ensure that runtimes are reasonable.
Figure 1b shows the probability distribution of the number of residues that lie within a SOAS of 10 or 11 Å) for all residues in proteins in our dataset. A residue Rj is considered to be within the SOAS of another residue Ri if any atom of Rj falls within a sphere of radius SOAS centered around the Cα of Ri. Out of a total of 174780 residues in these proteins, the maximum number of residues found within the SOAS of 10 Å is 58. Thus, the upper bound on the number of possible combinations for one residue is 4C58 = 424270, which is quite tractable (as can be seen from runtimes below). Note, that although the algorithm is polynomial in complexity, this number increases rapidly with increasing motif length, as well as the SOAS. For example, for a motif length of 5, the number of possible combinations in a set of 58 residues is 4582116 (ten times the number for a motif length of 4). However, a 4 residue motif is sufficient to represent most active site conformations, and for a preliminary search on extensive datasets. Similarly, for a SOAS of 11 Å we obtain the maximum number of residues as 70 - which results in 4C70 (= 916895) possible combinations (twice the number for a SOAS of 10 Å).
Running CLASP using the PREMONITION modified algorithm
We queried trypsin (PDBid:1A0J) with all the 500 motifs using the modified CLASP algorithm using the PREMONITION database19. The best matches are shown in Table 2. As expected, the best matches are those with known serine catalytic triads. As an illustrative example, we chose a protein (thioesterase - PDBid:1THT) with no known relationship with trypsin. This protein has the active site motif - H241 S114 V136 W213. This corresponds to the string query ‘HSVW’ (note that the string is sorted). We extract all entries for this string (Equation 5), which are all possible occurrences of the structural motif ‘HSVW’ in the protein.
Table 2. Best matches when trypsin (PDBid:1A0J) is queried using 500 motifs from the CSA database.
As expected, the best matches are those with known catalytic triads.
| PDB | Length | Description | CLASP Score |
|---|
| 1A0J | 223 | Trypsin | 0 |
| 1SSX | 198 | Alpha-lytic protease | 0.2 |
| 1AZW | 313 | Proline iminopeptidase | 0.7 |
| 1MEK | 120 | Protein disulfide isomerase | 1 |
| 2LPR | 198 | Alpha-lytic protease | 1.2 |
| 1SCA | 274 | Subtilisin carlsberg | 1.2 |
| 1C4X | 285 | 2-Hydroxy-6-oxo-6-phenylhexa-2,4-die | 1.3 |
| 1A7U | 277 | Chloroperoxidase T | 1.3 |
| 1LJL | 131 | Arsenate reductase | 1.3 |
| 1QJ4 | 257 | Hydroxynitrile lyase | 1.3 |
| 1RGQ | 200 | NS3 Protease | 1.3 |
| 1JKM | 361 | Esterase | 1.4 |
| 1EH5 | 279 | Palmitoyl protein thioesterase 1 | 1.4 |
| 2AAT | 396 | Aspartate aminotransferase | 1.4 |
| 1THT | 305 | Thioesterase | 1.5 |
= [(71, 26, 121, 141), (71, 26, 138, 141), (57, 214, 212, 215)...] (5)
All entries of ‘HSVW’ are compared using CLASP. Table 3 shows the electrostatic potential difference and spatial difference in each of the motifs in Equation 5. The best scoring motif is (H57ND1,S195OG,V213N,W215NE1). However, even the best motif has a relatively large RMSD (Figure 2). Thus, this is not a significant match.
Table 3. Potential and spatial congruence of the motif (H241 S114 V136 W213) from a thioesterase (PDB:1THT) in a trypsin protein (PDB:1A0J).
This motif corresponds to the key “HSVW”. D = Pairwise distance in Å. PD = Pairwise potential difference. APBS writes out the electrostatic potential in dimensionless units of kT/e where k is Boltzmann’s constant, T is the temperature in K and e is the charge of an electron.
| PDB | Active site atoms
(a,b,c,d) | | ab | ac | ad | bc | bd | cd |
|---|
| 1THT | H241ND1,S114OG,
VAL136N,W213NE1, | D
PD | 4.7
17.0 | 6.1
-97.0 | 6.3
-26.5 | 7.2
-114.0 | 9.0
-43.5 | 12.3
70.5 |
| 1A0J | H71ND1,S26OG,
VAL121N,W141NE1, | D
PD | 7.1
-288.7 | 16.3
-334.9 | 6.4
-227.3 | 13.4
-46.2 | 11.9
61.4 | 17.5
107.6 |
H71ND1,S26OG,
VAL138N,W141NE1, | D
PD | 7.1
-288.7 | 13.4
-346.2 | 6.4
-227.3 | 10.1
-57.5 | 11.9
61.4 | 13.5
118.9 |
H57ND1,S195OG,
V212N,W215NE1, | D
PD | 4.8
-20.7 | 7.9
-130.7 | 7.9
-52.3 | 6.7
-110.0 | 9.6
-31.6 | 9.7
78.4 |
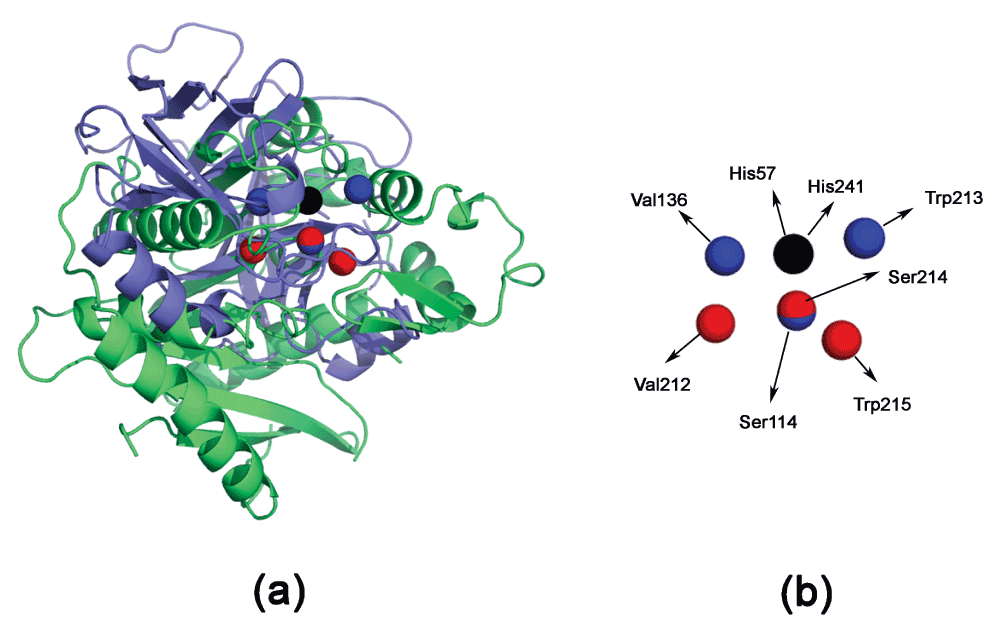
Figure 2. Superimposing thioesterase (PDBid:1THT, in blue) and trypsin (PDBid:1A0J, in green).
The proteins are superimposed based on the matching active site residues using DECAAF28. (a) The global superimposition does not show any significant homology. (b) The detailed residue configuration. Residues from trypsin are in red, and those in the thioesterase are in blue. His57 and His241 completely overlap and are in black.
Runtimes and disk space
Previously, we had proposed a computational method (PROMISE) to estimate the promiscuity of proteins with known active site residues and 3D structure using the CSA database25. PROMISE used each of the 500 proteins with known active site residues extracted from the CSA database to query every other protein in that set. This procedure required ~500*500=250000 program calls of CLASP19, each of which took one minute on an average. Thus, the total time taken was a month on a parallel system25. Using PREMONITION, it took less than a minute to query one protein with all 500 motifs. Thus, it took less than a day to replicate the PROMISE results. The precompilation step of extracting all motifs takes approximately 15 minutes on average for a single protein. For the protein with the largest SOAS (PDBid:1GPJ - 13 Å), the precompilation took one hour. These are acceptable values for a one time processing. The disk space for one protein PREMONITION file (zipped) is 14MB on average (7GB for 500 proteins).
Author contributions
SC wrote the computer programs. All authors analyzed the data, and contributed equally to the writing and subsequent revisions of the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant. BA acknowledges financial support from the Science Institute of the University of Iceland.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Bernstein FC, Koetzle TF, Williams GJ, et al.:
The Protein Data Bank: a computer-based archival file for macromolecular structures.
J Mol Biol.
1977; 112(3): 535–542. PubMed Abstract
| Publisher Full Text
- 2.
Altschul SF, Madden TL, Schaffer AA, et al.:
Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.
Nucleic Acids Res.
1997; 25(17): 3389–3402. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 3.
Gherardini PF, Wass MN, Helmer-Citterich M, et al.:
Convergent evolution of enzyme active sites is not a rare phenomenon.
J Mol Biol.
2007; 372(3): 817–845. PubMed Abstract
| Publisher Full Text
- 4.
Doolittle RF:
Convergent evolution: the need to be explicit.
Trends Biochem Sci.
1994; 19(1): 15–18. PubMed Abstract
| Publisher Full Text
- 5.
Rawlings ND, Barrett AJ:
Evolutionary families of peptidases.
Biochem J.
1993; 290(Pt 1): 205–218. PubMed Abstract
| Free Full Text
- 6.
Nadzirin N, Firdaus-Raih M:
Proteins of Unknown Function in the Protein Data Bank (PDB): An Inventory of True Uncharacterized Proteins and Computational Tools for Their Analysis.
Int J Mol Sci.
2012; 13(10): 12761–12772. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Russell RB:
Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution.
J Mol Biol.
1998; 279(5): 1211–1227. PubMed Abstract
| Publisher Full Text
- 8.
Kleywegt GJ:
Recognition of spatial motifs in protein structures.
J Mol Biol.
1999; 285(4): 1887–1897. PubMed Abstract
| Publisher Full Text
- 9.
Konc J, Janezic D:
Binding site comparison for function prediction and pharmaceutical discovery.
Curr Opin Struct Biol.
2014; 25: 34–39. PubMed Abstract
| Publisher Full Text
- 10.
Debret G, Martel A, Cuniasse P:
RASMOT-3D PRO: a 3D motif search webserver.
Nucleic Acids Res.
2009; 37(Web Server issue): W459–464. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Shatsky M, Shulman-Peleg A, Nussinov R, et al.:
The multiple common point set problem and its application to molecule binding pattern detection.
J Comput Biol.
2006; 13(2): 407–428. PubMed Abstract
| Publisher Full Text
- 12.
Bauer RA, Bourne PE, Formella A, et al.:
Superimpose: a 3D structural superposition server.
Nucleic Acids Res.
2008; 36(Web Server issue): W47–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Goyal K, Mohanty D, Mande SC:
PAR-3D: a server to predict protein active site residues.
Nucleic Acids Res.
2007; 35(Web Server issue): W503–505. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
Kirshner DA, Nilmeier JP, Lightstone FC:
Catalytic site identification--a web server to identify catalytic site structural matches throughout PDB.
Nucleic Acids Res.
2013; 41(Web Server issue): W256–265. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 15.
Konc J, Janezic D:
ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment.
Bioinformatics.
2010; 26(6): 1160–1168. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Holm L, Kaariainen S, Rosenstrom P, et al.:
Searching protein structure databases with DaliLite v.3.
Bioinformatics.
2008; 24(23): 2780–2781. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Angaran S, Bock ME, Garutti C, et al.:
MolLoc: a web tool for the local structural alignment of molecular surfaces.
Nucleic Acids Res.
2009; 37(Web Server issue): W565–570. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Shulman-Peleg A, Shatsky M, Nussinov R, et al.:
MultiBind and MAPPIS: webservers for multiple alignment of protein 3D-binding sites and their interactions.
Nucleic Acids Res.
2008; 36(Web server issue): W260– 264. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 19.
Chakraborty S, Minda R, Salaye L, et al.:
Active site detection by spatial conformity and electrostatic analysis--unravelling a proteolytic function in shrimp alkaline phosphatase.
PLoS One.
2011; 6(12): e28470. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Chakraborty S, Ásgeirsson B, Minda R, et al.:
Inhibition of a cold-active alkaline phosphatase by imipenem revealed by in silico modeling of metallo-β-lactamase active sites.
FEBS Lett.
2012; 586(20): 3710–3715. PubMed Abstract
| Publisher Full Text
- 21.
Rendon-Ramirez A, Shukla M, Oda M, et al.:
A computational module assembled from different protease family motifs identifies PI PLC from Bacillus cereus as a putative prolyl peptidase with a serine protease scaffold.
PLoS One.
2013; 8(8): e70923. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Chakraborty S, Rendon-Ramirez A, Ásgeirsson B, et al.:
Dipeptidyl peptidase-iv inhibitors used in type-2 diabetes inhibit a phospholipase c: a case of promiscuous scaffolds in proteins [v1; ref status: approved 1, approved with reservations 1, http://f1000r.es/2hw].
F1000Research.
2013; 2: 286. Reference Source
- 23.
Jaroszewski L, Li Z, Krishna SS, et al.:
Exploration of uncharted regions of the protein universe.
PLoS Biol.
2009; 7(8): e1000205. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Porter CT, Bartlett GJ, Thornton JM:
The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data.
Nucleic Acids Res.
2004; 32(Database): D129–133. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Chakraborty S, Rao BJ:
A measure of the promiscuity of proteins and characteristics of residues in the vicinity of the catalytic site that regulate promiscuity.
PLoS One.
2012; 7(2): e32011. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 26.
Baker NA, Sept D, Joseph S, et al.:
Electrostatics of nanosystems: application to microtubules and the ribosome.
Proc Natl Acad Sci U S A.
2001; 98(18): 10037–10041. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Dolinsky TJ, Nielsen JE, McCammon JA, et al.:
PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations.
Nucleic Acids Res.
2004; 32(Web Server issue): W665–667. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Chakraborty S:
An automated flow for directed evolution based on detection of promiscuous scaffolds using spatial and electrostatic properties of catalytic residues.
PLoS One.
2012; 7(7): e40408. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)