Keywords
microRNA, degradome, PARE, mRNA cleavage, Glycine max, Arabidopsis thaliana, qPCR, qACE
microRNA, degradome, PARE, mRNA cleavage, Glycine max, Arabidopsis thaliana, qPCR, qACE
Two of the reviewers pointed out the need for independent validation of change in levels of cleavage assayed by qACE. In response to these comments, we used published Northern blot data to quantify band intensities of full length and cleaved AtNAC1. In agreement with our results in soybean, miR164 over-expressing Arabidopsis seedlings had increased ratio of cleaved products while mir164 mutants of Arabidopsis had a reduced ratio of cleaved products. We have also modified generalized statements in our conclusions about “differential cleavage” and reorganized the manuscript in order to introduce qACE primer design, ratio of cleaved transcripts (RCT), and assumptions about stability of cleaved transcripts along with the results. We have specifically reiterated that qACE can only be used to assay previously validated targets where the cleavage site is known.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
miRNAs are short 21-22nt RNA molecules that regulate the expression of cognate target genes primarily through post-transcriptional mechanisms1. The majority of plant miRNAs fine-tune target gene expression through precise mRNA cleavage to achieve proper spatio-temporal expression patterns during plant development as well as in response to environmental changes2–4. Adaxial localization of transcripts encoding a set of HD-ZIP III transcription factors by miR166 in leaves5,6 is an excellent example. The expression of miR166 is restricted to abaxial layers in contrast to its target genes7. Mutations in miRNA recognition sites of HD-ZIP III genes (conferring resistance against miR166-mediated decay) resulted in ectopic expression of these genes in abaxial cell layers leading to adaxialized leaves8. Polarized expression of miR166 and its target HD-ZIP IIIs is conserved in other plant species as well, e.g. maize7, and soybean9. Another example is the nodule-meristem specific expression of MtHAP2-1 (alpha subunit of a CCAAT-binding NFY), which is spatially restricted by miR169, which is expressed in the infection zone adjacent to the meristem. Expression of a miRNA resistant MtHAP2-1 under the control of its native promoter led to significantly reduced nodule growth suggesting that spatially restricted expression of MtHAP2-1 is crucial for proper nodule development10. Similarly, miR399 plays an important role during inorganic phosphate (Pi) starvation by down regulating PHO2/UBC24 (a gene encoding a putative ubiquitin conjugating enzyme) mRNA11,12. PHO2 maintains proper levels of phosphate transporter activities12 and its down regulation increases phosphate acquisition. These examples demonstrate that mere transcriptional regulation is not sufficient to achieve proper spatial and temporal expression of target genes and miRNA regulation is crucial for additional fine-tuning of gene expression.
Identification, validation and quantification of the extent of regulation of specific target genes by miRNA is crucial for proper understanding of miRNA-mediated gene regulation. Bioinformatics-based prediction of miRNA targets was developed based on the extent of base-pairing between miRNAs and their targets13 and subsequently improved based on base-pairing in specific “seed” regions as well as the cleavage site11,14. A defining feature of miRNA-guided regulation in plants is that cleavage takes place precisely between 10th and 11th nucleotide from the 5’end of miRNA in the complementary region of the target transcript. A modified RNA Ligase Mediated-5’Rapid Amplification of cDNA Ends (RLM-5’RACE) method is being used to validate cleavage by miRNAs by mapping the site of cleavage15. In this technique an RNA oligo of known sequence is ligated to the mRNA molecules that had an uncapped 5’end arising due to RNase cleavage. OligodT-primed cDNA molecules from adapter ligated RNA is then used to amplify specific target RNA molecules (using a forward primer from the adapter and gene-specific reverse primer). Subsequent sequencing of these amplicons can precisely map the cleavage site. This technique serves as an excellent qualitative method to validate miRNA-mediated cleavage of targets. However, this method cannot quantify the extent of miRNA cleavage i.e. increased cleavage in specific tissue types and/or specific developmental stages/time points. In addition, among the multiple targets of a particular miRNA, some are cleaved more efficiently than the others for example, miR393 mediated reduction in the flag22 elicited wild type seedlings, showed reduction of TIR1, AFB2, AFB3 but not AFB116. Recently, high throughput sequencing-enabled methods were developed for genome-wide validation (and identification) of miRNA targets (“degradome” or PARE (Parallel Analysis of RNA Ends))17,18. Briefly, an RNA adaptor with a MmeI recognition site is ligated to the cleaved ends of polyA RNA molecules. Ligated molecules are reverse-transcribed, amplified linearly and digested with MmeI (which cleaves ~20 nucleotides (nt) away from the recognition site) yielding short DNA molecules corresponding to the junction of the RNA adapter and miRNA-cleavage end. A dsDNA oligo adapter is subsequently ligated to the MmeI end and these molecules are linearly amplified, and sequenced using high throughput methods. This results in ~20 nt cleavage signatures that can be used to map miRNA-directed cleavage sites (e.g. CleaveLand bioinformatics pipeline (Addo-Quaye et al. 2009). Normalized abundance of cleavage signatures from specific target genes can be used to quantify miRNA-directed cleavage. However, miRNA binding sites of target genes are highly conserved. Therefore, these short signature sequences generated by degradome/PARE analyses might be shared by more than one gene making it impossible to distinguish cleavage levels of specific target transcripts. For example, at least two targets of miR319 share the same cleavage signature in Arabidopsis (Table S1). Other examples include targets of miR161 and miR172. This is especially significant in species with extensive genome duplications (e.g. soybean) and polyploidy species (e.g. wheat) that have a number of paralogs with high sequence identity. For instance, in soybean five auxin response factor (ARF) genes targeted by miR160 shared the same cleavage signature in the degradome library and RLM-5’RACE had to be used to confirm cleavage of individual target genes19. In addition, degradome/PARE analysis is a global analysis method and does not allow determining cleavage levels of a small set of miRNA targets (Table S1). A semi-quantitative method to assay miRNA-directed cleavage was reported by Schwab et al. (2005)14. However, the method was not widely used subsequently perhaps due to the lack of validation under different conditions and/or its semi-quantitative nature. We enhanced this method through the use of qPCR and named it “quantitative Amplification of Cleaved Ends” (qACE), and evaluated its ability to assay the extent of miRNA-directed cleavage of specific target genes. qACE allows quantification of specific target mRNA molecules resulting from miRNA-directed cleavage in different tissue-types and/or under different experimental conditions. We demonstrate the applicability of this method to (i) assay increased cleavage of targets in miRNA over-expressing plants, (ii) assay reduced cleavage of targets in miRNA-deficient plants, and (iii) to identify a key role for miR164 in conferring nodule-enriched expression of two of its target genes.
Arabidopsis thaliana wild type Ler and hen1-1 mutant seeds (obtained from ABRC, Columbus, OH: Stock#CS6583) were surface-sterilized and grown in Sunshine mix #1 (Tessman Company, Sioux Falls, SD) under 16h light at 25°C. Leaf, stem and flower tissues were harvested at the same developmental stages, immediately frozen in liquid N2 and stored at -70°C. Soybean seeds Glycine max cv. Williams-82, the genotype used for genome sequencing project20 were surface-sterilized21 and germinated in 4” pots filled with a mixture of vermiculite: perlite (Hummert International, MO) in the ratio of 1:3 and watered with nitrogen free plant nutrient solution22. The plants were grown in a controlled environment vertical growth chamber (Conviron Growth chamber, Manitoba, Canada). Growth conditions used were: 16h light and 8h dark and 50% relative humidity with a day and night temperature of 25°C and 20°C respectively. For nodulation assays 5 days old germinated plants were inoculated with Bradyrhizobium japonicum (USDA110) grown in Vincent’s rich medium prepared and supplemented with Chloramphenicol (antibiotic selection marker) at 30°C23. The plants were inoculated with B. japonicum at a concentration of OD600=0.08 and 14 days post inoculation mature nodules and the root segment adjacent to the nodule were harvested separately.
To over-express miR164, the precursor of gma-miR164 (miRBaseID MI0007209) was PCR amplified using G. max genomic DNA as template. The PCR product was initially cloned in to an entry vector PCR8/GW/TOPO (Invitrogen, Carlsbad, CA) and subsequently in to the binary vector pCAMGFP-CsVMV: GW24 using a Gateway LR clonase (Invitrogen, Carlsbad, CA) reaction. The vector was transformed in to Agrobacterium rhizogenes K599 cells, using electroporation and composite plant transformation was performed as described25. Transgenic GFP roots were collected on dry-ice and stored at -70°C until RNA isolation and cDNA synthesis.
Total RNA was isolated from the different tissues using TRI reagent (Product#T9424, Sigma Aldrich, St. Louis, MO) as described previously21. The RNA was quantified using Nanodrop spectrophotometer (ND1000, Thermo Scientific, Wilmington, DE) and RNA integrity was verified using agarose gel electrophoresis. Adapter ligated cDNA was prepared using GeneRacer kit (Product#L1502-01, Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions except that calf intestinal phosphatase treatment to remove 5’ phosphate and 5’ Cap removal using tobacco acid pyrophosphatase steps were omitted. For Arabidopsis gene expression analysis 2µg of total RNA each of leaf, stem, and flower was pooled together and subject to adapter-ligated cDNA synthesis. For gene expression analysis in soybean nodules and adjacent root tissues, 7µg of total RNA was used in adapter-ligated cDNA synthesis. For miRNA stem-loop cDNA synthesis, 1µg of total RNA was used and a multiplexed cDNA was performed for all miRNAs examined using M-MuLV reverse transcriptase26,27. miR1515 & U6 was used as normalization/house-keeping control for miRNAs28.
qPCR assays were performed in MX3000P thermocycler (Stratagene/Agilent technologies, Santa Clara, CA) using SYBR Advantage qPCR premix (Product#639676, Clontech, Mountain View, CA). The data was analyzed using the MxPro software. Relative expression values for full length transcripts were performed using the dCt method29 using Actin for soybean and U6 for Arabidopsis as normalization control. Relative levels of cleaved transcripts (described as RCT in text) were calculated using the dCt method comparing the Ct values of full-length vs. cleaved transcripts in the same sample. For statistical analyses, we calculated the range of possible expression values in each sample based on the deviation between Ct values of replicates. Error bars indicate this range in each sample.
qACE is an extension of the modified RLM-5’RACE method15,30 used to qualitatively validate target cleavage and the subsequent semi-quantitative method14 used to examine the levels of cleavage remnants. An RNA adapter is ligated to cleaved 5’-ends of polyA or total RNA preparations (“adapter-ligated RNA”). The adapter gets ligated to any available 5’-phosphate of ribose molecule of RNA arising due to miRNA-directed cleavage or other means (e.g. mRNA degradation), but not to full-length mRNAs, because of the 5-methyl Guanosine/CAP. Oligo-dT primed cDNAs generated from adapter-ligated RNA are subject to real-time PCR where a primer that spans the adapter junction is used as a forward primer (subsequently referred to as qACE forward primer) and a gene-specific reverse primer (Figure 1).
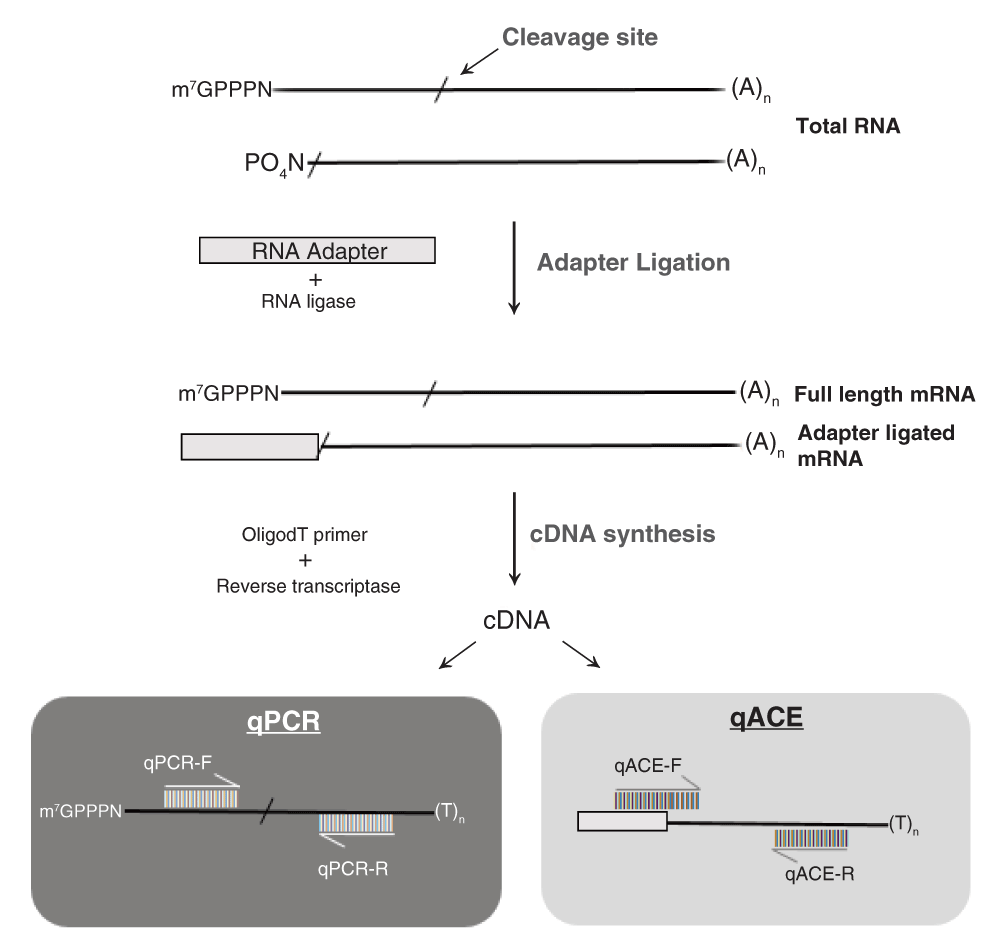
Total RNA containing full length transcripts with 7-methyl Guanosine/CAP and cleaved transcripts arising due to miRNA activity is used as starting material. An RNA oligo adapter of known sequence is ligated to the cleaved transcripts (with a 5’-Phosphate group) giving rise to adapter ligated mRNA. Oligo dT-primed cDNA is used to assay the levels of full length mRNA (qPCR) and cleaved transcripts (qACE). The qPCR forward and reverse primers (qPCR-F & qPCR-R) were designed across the miRNA binding site whereas the qACE forward primer (qACE-F) is designed complementary to adapter sequence but with six nucleotides at the 3’end specific to the sequence at 5’end of cleaved transcript, and a gene specific reverse primer (qACE-R).
The forward primer had to be designed such that it does not amplify full-length cDNA molecules or bind non-specifically to other adapter-ligated molecules. We achieved this by careful design such that this primer corresponds to the adapter-cleavage product junction with 6 nucleotides at the 3’end of the primer corresponding to the nucleotides downstream of the cleavage site in the target gene. A shorter design (a 4nt sequence for example), had an increased probability of non-specific binding. On the other hand, longer than 6nt design had a higher thermodynamic stability making it possible to bind to full-length cDNA molecules (depending on the GC content). In addition, for target genes with identical miRNA cleavage signatures (and therefore identical qACE forward primers), specificity was achieved by carefully designing gene-specific reverse primers.
We expected that the qACE forward primer combined with a gene-specific reverse primer would help specifically amplify ligation products where the adapter is ligated to the predicted/validated cleavage site. Therefore, a qPCR assay using these primers will specifically quantify cleaved mRNAs resulting from miRNA-directed cleavage. We also expected that the qACE forward primer will not amplify full-length cDNA molecules and neither would it efficiently amplify ligation products where the cleaved end does not correspond to the miRNA-directed cleavage site (Figure S4a1). Finally, we also expected that the use of a gene-specific reverse primer would amplify linearly and help distinguish different targets of the same miRNA even if the miRNA-binding sites are identical/highly conserved (Figure S4a–Figure S4d). In addition, the same cDNA preparation can be used to quantify full-length molecules of target mRNA as well as using appropriate primers that span across the miRNA binding site (subsequently referred to as full-length qPCR primers). Ratio of cleaved transcripts vs. full-length transcripts will serve as a quantitative indicator of the extent of miRNA-directed cleavage of the target mRNA. The level of cleaved target transcripts also depends on the levels of transcriptional activity of the target gene. Therefore, comparing absolute levels of cleaved transcripts (or levels normalized to house-keeping genes) between two conditions or tissue types might not reveal differences in miRNA regulation. We decided to normalize the levels of cleaved transcripts to full-length transcripts using the dCt method to obtain the ratio of cleaved transcripts (RCT). The formula for calculating RCT is, 2(Ct(Full length)-Ct(cleaved transcript)), where Ct stands for threshold cycle. It should be noted that qACE relies on prior knowledge about the validity of the cleavage site e.g. obtained through 5’-RACE or degradome/PARE analyses. It cannot be used to validate a predicted cleavage site. In addition, one has to assume that the stability of cleavage products are identical among multiple target genes that are compared. However, such an assumption has to be made even for alternate methods such as degradome/PARE analysis.
To obtain proof of concept for the method, we examined the abundance of cleavage remnants in miR164 over-expressing soybean roots. We isolated total RNA from vector control and miR164 over-expressing roots and examined mature miR164 levels by stem-loop qPCR. We observed an 6-fold increase in miR164 over-expressing roots compared to control roots as expected (Figure 2a)31. We also examined the expression of GmNAC1b, a 5’-RACE-validated target of miR164 in soybean (Figure S1). GmNAC1b is a close ortholog of AtNAC1 (Figure S2). A potential cleavage product of AtNAC1 accumulates in the roots of Arabidopsis plants over-expressing miR16433. To quantify the levels of GmNAC1b, we generated oligo-dT-primed cDNA and performed qPCR assays using primers designed across the miR164 binding site of GmNAC1b. Results from qPCR assays indicated that there was a 4.2-fold reduction in the levels of full-length GmNAC1b transcripts in miR164 over-expressing roots compared to control roots (Figure 2b). These results indicated that indeed over-expression of miR164 resulted in a reduction in full-length transcripts of its target.
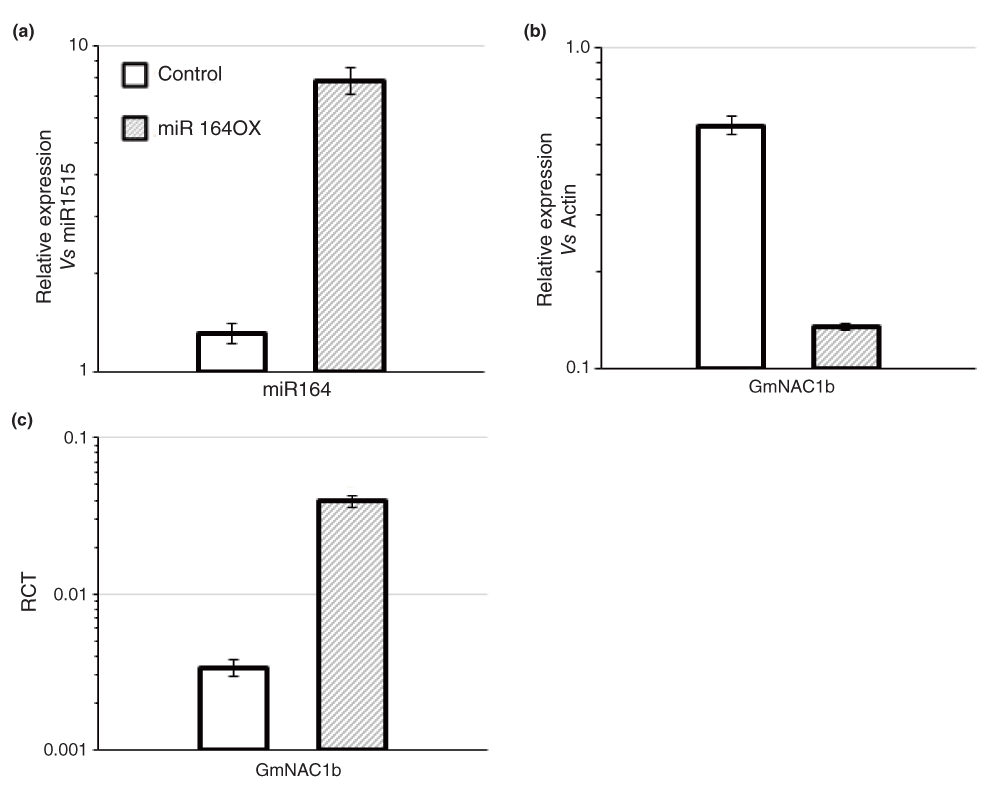
(a) The relative expression level of miR164 (normalized to that of miR1515) in control soybean roots and those over-expressing miR164 (miR164ox) determined using stem-loop qPCR. (b) The relative expression levels of full-length GmNAC1b (normalized to that of GmActin) in control and miR164ox roots assayed using qPCR. The data shown in (a) and (b) are average of three replicate assays and error bars indicate the range of possible values based on deviation between Ct values of replicate assays. (c) The Ratio of Cleaved Transcripts (RCT) over full-length transcripts of GmNAC1b in control and miR164ox roots assayed using qACE. The data shown are average of three replicate assays and error bars indicate the range of possible values based on deviation between Ct values of three replicate assays.
Next, we used qACE to detect and quantify target cleavage in these roots. We designed qACE primers for GmNAC1b as outlined in Figure 1. First we examined if the qACE primer pair does not amplify full-length molecules of GmNAC1b using the above cDNA from total RNA without any adapter ligation. qPCR assays detected no amplification (Figure S4a1) indicating that indeed qACE primers did not amplify/detect full-length GmNAC1b molecules. Next, we prepared adapter-ligated cDNA by ligating a known RNA adapter (see methods) to total RNA and reverse-transcribing these molecules using an oligo-dT primer. We used this cDNA to assay full length and cleaved molecules of GmNAC1b using the appropriate primer pairs (Figure 1) and calculated RCT values for each NAC1b in control and miR164 over-expressing roots. There was an 11.6-fold increase in the ratio of cleaved GmNAC1b transcripts in miR164 over-expressing roots compared to the control roots (Figure 2c) indicating that indeed miR164 over-expressing roots had increased cleavage of GmNAC1b. This experiment demonstrated that qACE could detect and provide a quantitative indicator of the levels of target cleavage.
Next, we examined the levels of target cleavage in Arabidopsis wild-type (Ler) and miRNA-deficient hen1-1 mutant plants. It was previously demonstrated using Northern blots that hen1-1 mutants accumulated less amounts of miRNAs and that there was an increase in the levels of miRNA targets in these plants34. We used stem-loop qPCR to determine the levels of three different conserved miRNAs: ath-miR160, ath-miR164 and ath-miR159 in Arabidopsis Ler and hen1-1 plants27. As reported earlier, a clear reduction in the levels of mature miR160, miR164 and miR159 was observed in hen1-1 plants in comparison to Ler (control) (Figure 3a). However, it should be noted that the levels of reduction were not uniform for all three miRNAs; fold change of -3.2, -21, and -18 respectively.
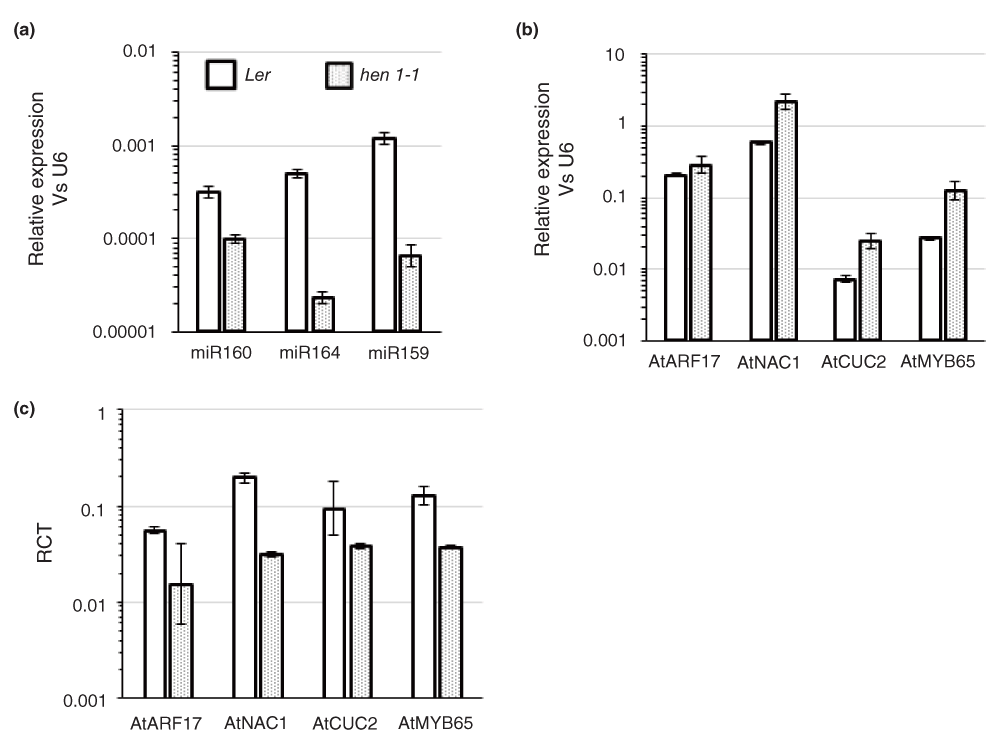
(a) Expression of ath-miR160, ath-miR164 and ath-miR159 in wild-type Ler and hen1-1 tissues assayed by stem-loop qPCR. The expression levels of the miRNAs were normalized to U6. The data shown are average and error bars indicate the range of possible values based on deviation between Ct values of three replicate assays. (b) Levels of full-length target transcripts (AtARF17 target of miR160, AtNAC1 & AtCUC2 target of miR164 and AtMYB65 target of miR159) in Ler and hen1-1 tissues analyzed using qPCR. Gene expression levels were normalized to U6 and further confirmed using two additional housekeeping genes, AtEF-α and AtGAPDH (data not shown). The data shown are average and error bars indicate the range of possible values based on deviation between Ct values of two replicate assays. (c) The Ratio of Cleaved Transcripts (RCT) over full-length transcripts of AtARF17, AtNAC1, AtCUC2, AtMYB65 in Ler and hen1-1 tissues assayed by qACE. Data shown are average and error bars indicate the range of possible values based on deviation between Ct values of two replicate assays.
Next, we used qPCR and qACE respectively to determine the levels of full length and cleaved transcripts of selected targets of the above miRNAs. AtARF17 is an auxin response factor post-transcriptionally regulated by ath-miR16032. As reported previously, we also observed an increase in the expression of AtARF17 in hen1-1 plants (~1.4-fold increase; Figure 3b). Similarly, AtNAC1 and AtCUC2, targets of ath-miR164 showed ~3.5 fold increase in gene expression levels in hen1-1 compared to Ler (Figure 3b). AtMYB65, known to be regulated by ath-miR159 had a 4.6-fold increase in transcript levels in hen1-1 compared to Ler (Figure 3b). qACE assays indicated a significant reduction in the abundance of miRNA-directed cleavage products for all these target genes in hen1-1. For example, the levels of cleaved AtARF17 levels were lower in hen1-1 plants resulting in a ~3.5-fold reduction in RCT (Figure 3c). Similarly, AtNAC1 had a 6.3-fold reduction in RCT in hen1-1 compared to Ler. AtCUC2 another target of miR164 had only a 2.5-fold decrease in RCT in hen1-1 vs. Ler. Note that both AtNAC1 and AtCUC2 had a 3.5-fold increase in the levels of full-length transcripts (hen1-1 vs. Ler) but the reduction in RCTs was very different. This data indicated that the extent of gene expression regulated by miR164 is perhaps different between these two targets. Possible reasons include differences in cleavage efficiency as well as differences in the extent of overlap in tissue domains where the miRNA and the targets are expressed. qACE helped identify this difference where as simple comparison of full-length transcript levels by qPCR would not have identified it. Finally, AtMYB65 showed a ~3.5-fold reduction in RCT in hen1-1 vs. Ler (Figure 3c).
As a comparison, we determined the ratio of cleaved vs. full length target transcripts using previously published northern blot data using Arabidopsis plants with increased or decreased levels of miR16433. We performed image analysis to obtain band intensities of full length and cleaved products and calculated the ratio of cleaved transcripts to full length. In Arabidopsis plants over-expressing miR164 in a chemically inducible manner, a 48 hour induction led to a 1.5 fold increase in RCT in one of the transgenic lines (Table S2). In the other transgenic line, the cleaved product was below the limit of detection at this time point and hence RCT could not be calculated. Nevertheless, increased RCT in miR164 over-expressing plants was consistent with results from our soybean miR164 over-expression experiments (Figure 2). Similarly, in mir164 mutant lines, we observed a significant decrease in RCT (Table S3) consistent with results from our hen1-1 experiments (Figure 3). Evidence from these experiments clearly demonstrated the ability of qACE to provide a quantitative indicator of miRNA-directed cleavage of targets resulting from changes in cognate miRNA levels.
Having demonstrated the ability of qACE to detect and quantify cleavage of specific miRNA targets, we used the technique to examine the role of miRNAs in governing nodule-specific/enriched gene expression in soybean. We compiled a total of 326 miRNA genes classified in to 134 families from soybean and predicted a total of 596 genes targeted by these miRNAs in soybean35. Among these, a set of miRNA-target pairs had inverse expression pattern between mature symbiotic nodules (MN) and adjacent root tissues (ABMN; data not shown). We identified two NAC transcription factors, GmNAC1a and GmNAC1b (5’-RACE validated targets of miR164) that were expressed in a nodule-enriched manner. While GmNAC1a had ~6.3-fold higher expression in MN vs. ABMN tissues, GmNAC1b had a 3.2-fold higher expression (Figure 4a). Interestingly, miR164 had a nodule-excluded expression pattern i.e. ~40-fold lower expression in MN vs. ABMN (Figure 4b).
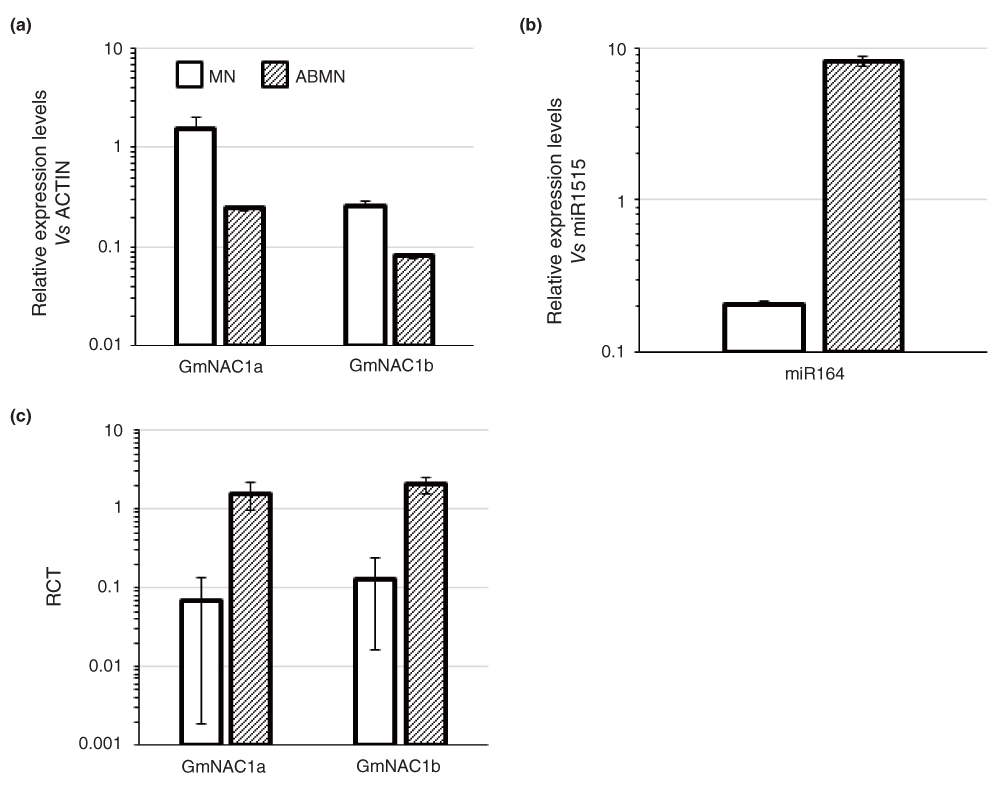
(a) Levels of full-length GmNAC1a & b transcript levels in mature nodules (MN) and adjacent root tissues (ABMN) assayed by qPCR and expression levels were normalized to GmActin. Data shown are average and error bars indicate the range of possible values based on deviation between Ct values of two replicate assays. (b) Level of gma-miR164 in MN and ABMN tissues assayed by stem-loop qPCR. miRNA expression levels were normalized to that of miR1515. Data shown are average and error bars indicate the range of possible values based on deviation between Ct values of three replicate assays. (c) The Ratio of Cleaved Transcripts (RCT) over full-length transcripts of GmNAC1a & b assayed by qACE. Data shown are average from two biological replicates and error bars indicate ± SD.
Based on such inverse expression of these miRNA-target pairs we hypothesized that nodule-specific expression of these target genes might be at least in part regulated by nodule-excluded expression of miR164. In other words, miR164 might actively cleave these target genes in adjacent root tissues to restrict their expression to the nodules. Such a mechanism combined with transcriptional regulation can be used to generate expression gradients as observed between miR166 and its HD-ZIP III proteins during xylem development36. However, mere inverse expression of miRNAs and their targets is not conclusive evidence for our hypothesis. If indeed, these target genes were preferentially cleaved in ABMN tissues to ensure MN-specific expression, we would expect to observe a larger proportion of cleaved transcripts in ABMN tissues vs. MN tissues.
We used qACE to determine the cleavage levels of GmNAC1a and GmNAC1b in ABMN and MN tissues and calculated RCT values. It is worth noting that these targets share the same cleavage signature in degradome/PARE analyses (Table S1). We designed gene specific reverse primers that distinguished these genes (Table S4 and Figure S3a & b). Both GmNAC1a and GmNAC1b had increased cleavage in ABMN tissues compared to MN tissues. Interestingly, consistent with the higher enrichment of GmNAC1a in MN tissues, this gene had a higher cleavage in ABMN tissues (~23.5-fold higher than MN tissues). GmNAC1b also had higher cleavage in ABMN tissues (~15.8-fold higher than MN tissues; Figure 4c). Increased cleavage of GmNAC1a in ABMN compared to GmNAC1b was consistent with increased enrichment of full length GmNAC1a transcripts in MN compared to that of GmNAC1b. qACE assays strongly support the hypothesis that increased cleavage of targets in ABMN tissues by miR164 contributes at least in part to determine their nodule-enriched expression. The high sequence identity between the transcripts of GmNAC1a and b (~92%) precluded the use of Northern to perform such comparisons between these genes.
In summary, we have demonstrated that qACE can detect and quantify miRNA-directed cleavage of specific target genes using miRNA over-expression and miRNA-deficient tissues. We successfully used the technique to identify differential cleavage of specific target genes between nodule and adjacent root tissues revealing an additional miRNA-directed mechanism that results in nodule-specific/enriched gene expression.
qACE provides a quantitative indicator of miRNA-directed cleavage of specific target genes by assaying the levels of cleavage remnants. A number of studies have shown reduced levels of target transcripts as an indirect evidence of miRNA-directed cleavage36,37. Northern hybridization has also been used to estimate miRNA-directed cleavage. For example, a putative cleavage product of NAC1 resulting from miR164 activity was detected using a 3’-specific probe in Arabidopsis33. More recently, degradome/PARE analysis has enabled quantification of global miRNA cleavage17,18. However, this method cannot distinguish target transcripts that share the same cleavage signature (see Introduction). qACE, an enhancement to the semi-quantitative method developed by Schwab et al. 200514, enables quantification of miRNA-directed cleavage of a smaller set of target genes and also can differentiate closely related genes. During the preparation of this manuscript a near identical method was used by Li et al. (2014)38 to distinguish efficiencies of miR159-directed cleavage of different MYB33 targets with specific mismatches. This study validated the usefulness of the qACE method. We have performed proof of concept and control experiments (e.g. demonstrating lack of amplification using qACE primers in non-adapter ligated cDNA preparations) to demonstrate the method’s utility. We have demonstrated this using GmNAC1a and GmNAC1b which share a similar cleavage signature (Table S1). Our results show that using qPCR (to assay reduction in full-length transcripts) and qACE (to assay the levels of cleaved transcripts) on the same cDNA preparation, one can estimate specific levels of cleavage for each target gene. For example, we demonstrate that both NAC1 and CUC2 had similar levels of increase in hen1-1, but different levels of reduction in miR164-directed cleavage.
A potential limitation of qACE is the level of target gene expression and thus the detectable level of cleavage products. Among the different genes we tested, RCTs ranged from 0.0033 (GmNAC1b Figure 2c) to 0.2 (AtNAC1; Figure 3c). As one can imagine, for very poorly expressed genes with a low levels of miRNA cleavage, it can be technically challenging to reliably detect cleavage products. We used linearity and efficiency assays to determine valid range of Ct values that are reliable for each gene of interest (See below). Alternate solutions to this deficiency include the use of xrn4 mutants as done previously18 or the use of linear pre-amplification of the adapter-ligated RNA molecules by incorporating a T7 promoter in the adapter. However, this will amplify only cleaved products and not full-length transcripts. In addition, specific miRNAs also induce generation of secondary sRNAs from the cleaved product making them unavailable for quantification. Another limitation of qACE is its inability to distinguish cleavage products with closely spaced cleavage sites. For example, G. max has twenty miR166 genes that are predicted to target all twelve GmHD-ZIP III genes. We have identified two major cleavage products from several of these genes where the position of cleavage site differs by two nucleotides. Based on the known miR166 variants in soybean, we hypothesized that these two cleavage products arose from the activities of miR166a and miR166h respectively (Figure 5). We attempted to distinguish these cleavage products by qACE. Unfortunately qACE failed to differentiate the cleaved molecules arising due to gma-miR166h owing to loop formation in template which enables amplification of the qACE primer pairs designed to detect cleavage levels arising due to miR166a. Despite these minor limitations, we have demonstrated that qACE is a very useful method that can provide a quantitative indicator of the cleavage levels of specific miRNA target genes.
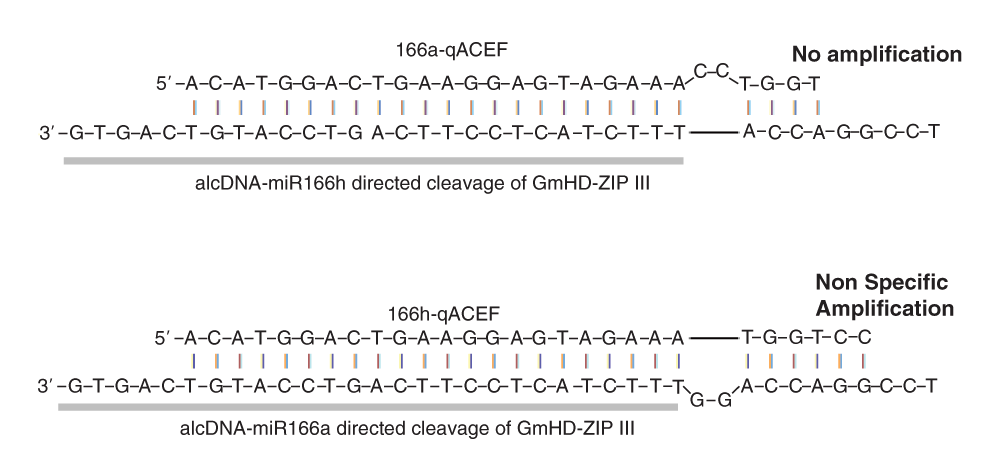
The images depict binding of the qACE forward primer (qACE-F) to cleavage products resulting from miR166h and miR166a guided cleavage of GmHD-ZIP III transcripts. The primer-template combination in the top panel resulted in no amplification underscoring the specificity of the primer. However, the primer-template combination shown in the bottom panel resulted in non-specific amplification. We hypothesize that the 6nt binding stretch at the 3’end was the reason for such non-specific amplification. The oligo adapter ligated to the cleaved end is indicated in bold letters and as grey bar.
qACE being a qPCR-based method, specificity and linearity of primers used are key factors to be considered39. For linearity assays for qACE primers, one has to use adapter-ligated cDNA. Due to the lower abundance of cleavage products, we made cDNA preparations from 5–7µg of DNase treated total RNA. To efficiently utilize these cDNA preparations for qACE assays, we used PCR amplified cDNA for linearity assays. We amplified a small amount of adapter-ligated cDNA using 5’adapter as forward primer and 3’PolydT adapter as reverse primer in a 25 cycle PCR. We reasoned that dilutions of this PCR product can be used to check the linearity of the qACE primer pairs, but cannot be used for qACE assays (due to non-linear amplification). This enabled the use of adapter-ligated cDNA primarily for qACE assays and increased the overall efficiency of the method in terms of amount of RNA requirement.
F1000Research: Dataset 1. Raw data qACE to assay miRNA-directed target cleavage, 10.5256/f1000research.5266.d3638340
SS conceived the study, guided data analysis, and wrote the manuscript. SD, SA and MT performed the experiments. SD and SA analyzed the data and co-wrote the manuscript.
Research in the authors’ laboratory is supported by funds (awarded to S.S) from USDA-AFRI (2010-65116-20514), NSF-PGRP (IOS-1350189), South Dakota Soybean Research and Promotion Council and South Dakota State University and Agricultural Experiment station.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The use of equipment at the SDSU-Functional Genomics Core Facility supported in part by NSF/EPSCoR Grant No. 0091948, the State of South Dakota is acknowledged.
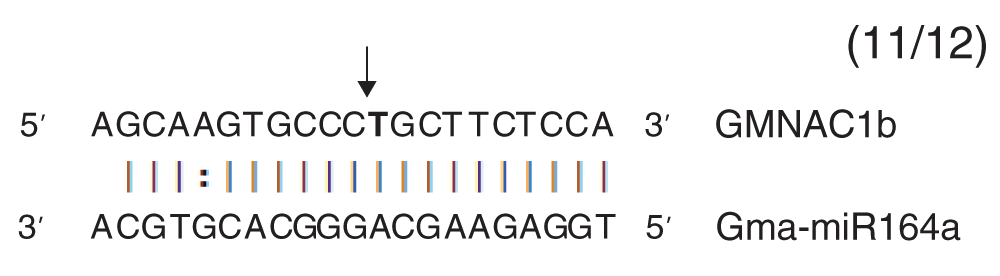
The arrow indicates the cleavage site in the miR164a binding site of GmNAC1b. The numbers within parenthesis indicate the frequency of miRNA cleaved product of the total number of sequences analyzed.
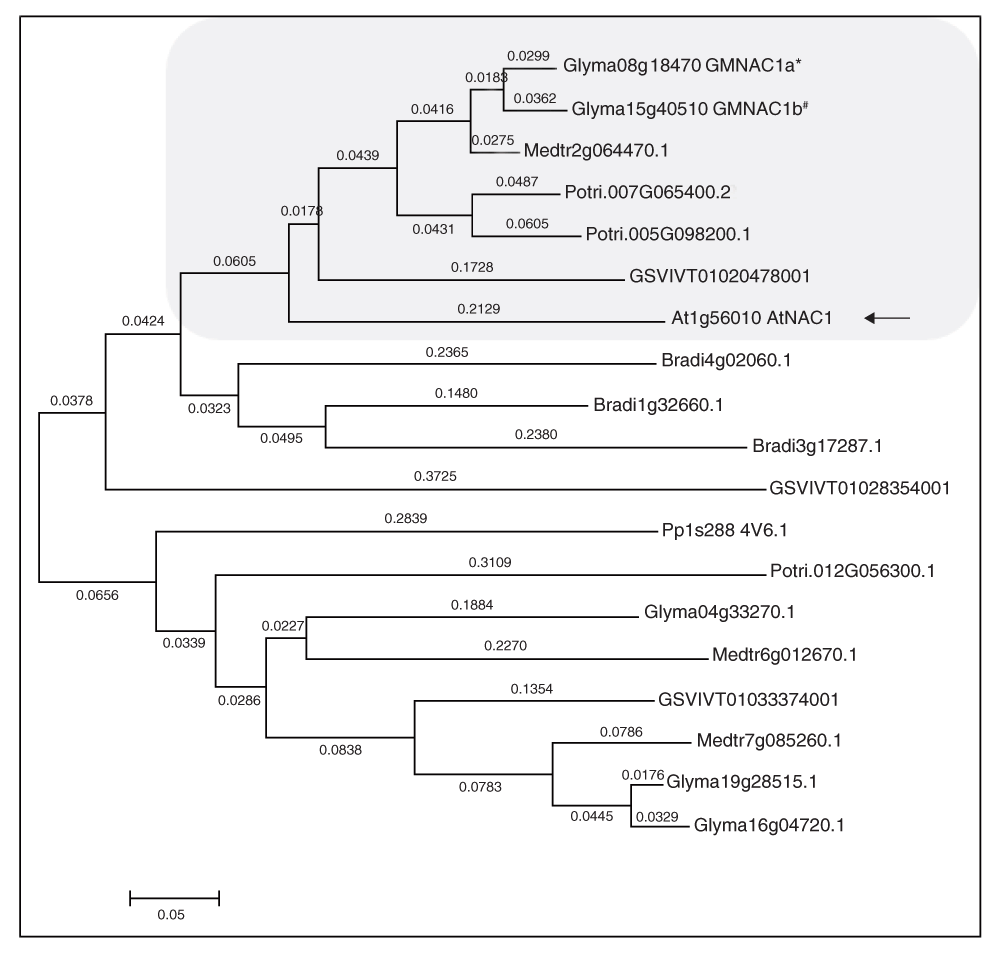
The amino acid sequences were aligned using global alignment in MEGA 5. The aligned sequences were used for constructing a phylogenetic tree using the rooted neighborhood joining method. The numbers in the branches indicate the distance and arrows shows the GmNAC1b presence in AtNAC1 clade.
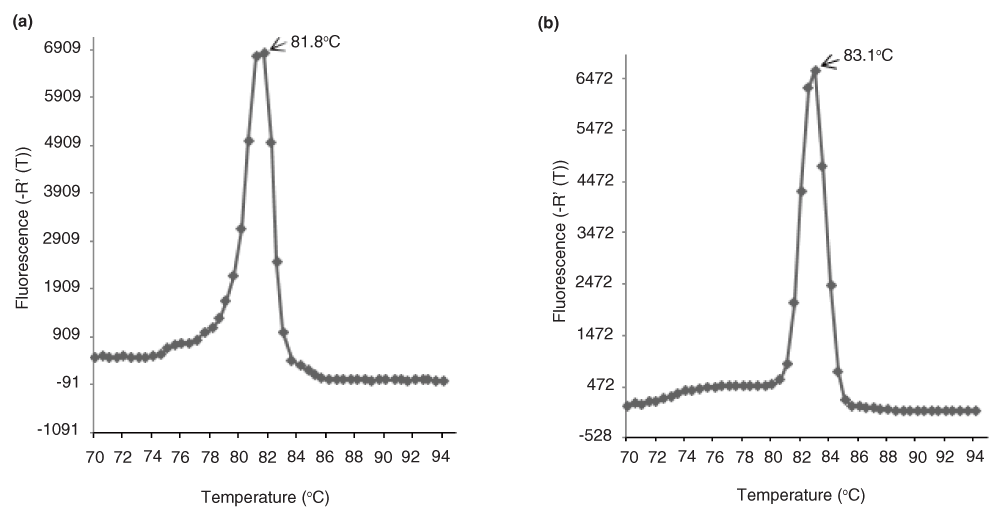
Dissociation curves of qACE amplicons (a) GmNAC1a and (b) GmNAC1b. The melting temperatures (Tm) are different for GmNAC1a and GmNAC1b underscoring the specificity of the gene specific reverse primer although the qACE-F primer is common for both. The data is the average of two replicate assays.
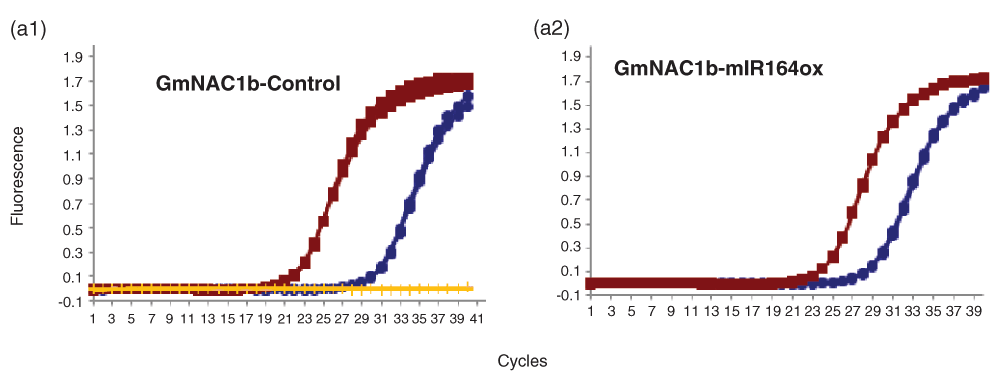
Amplification plots of GmNAC1b in (a1) control and (a2) miR164ox (miR164 over expression) amplification plots showing linear amplification of full length (red squares) and cleaved cDNA (blue circles) by GmNAC1b qPCR-F & R and qACE-F & R primers respectively determined by SYBR fluorescence. (a1) Amplification plot showing no amplification of full length cDNA by GmNAC1b qACE-F & R primers determined by SYBR fluorescence (yellow lines). The data shown are normalized fluorescence from three replicate assays.
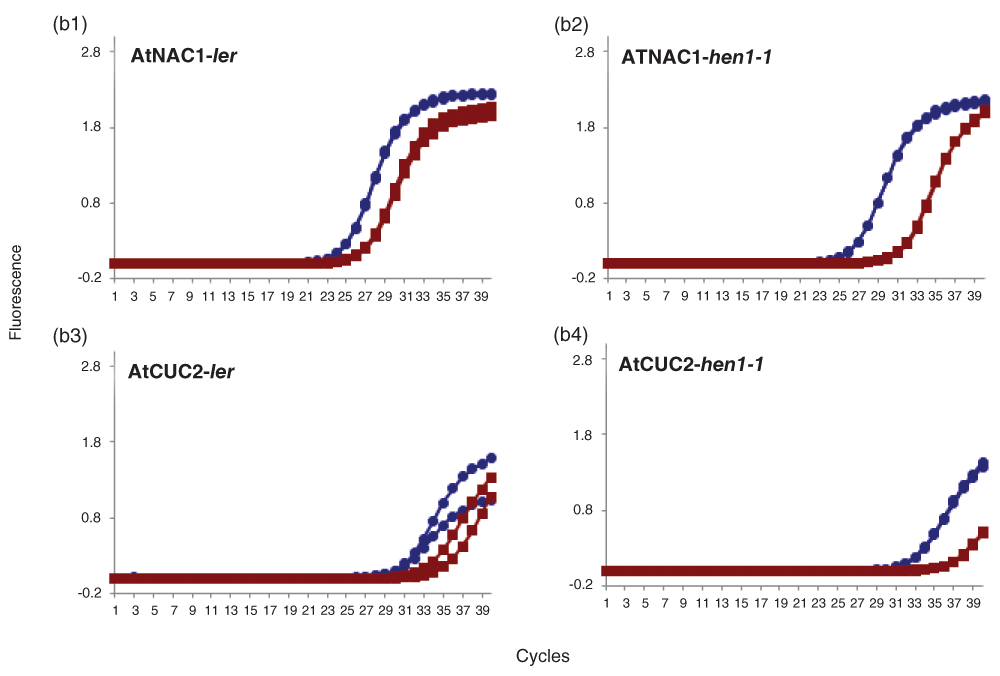
Amplification plots of AtNAC1 & AtCUC2 in (b1 & b3) Ler and (b2 & b4) hen1-1 Arabidopsis mutant. Amplification plot showing linear amplification of full length (blue circles) and cleaved cDNA (red squares) by qPCR-F & R and qACE-F & R primers respectively for AtNAC1 (b1 & b2), AtCUC2 (b3 & b4) determined by SYBR fluorescence. The data shown are normalized fluorescence from two replicate assays.
Amplification plots of AtARF17 & AtMYB65 in (c1 & c3) Ler and (c2 & c4) hen1-1 Arabidopsis mutant. Amplification plot showing linear amplification of full length (blue circles) and cleaved cDNA (red squares) by qPCR-F & R and qACE-F & R primers respectively for AtARF17 (c1 & c2), AtMYB65 (c3 & c4) determined by SYBR fluorescence. The data shown are normalized fluorescence from two replicate assays.
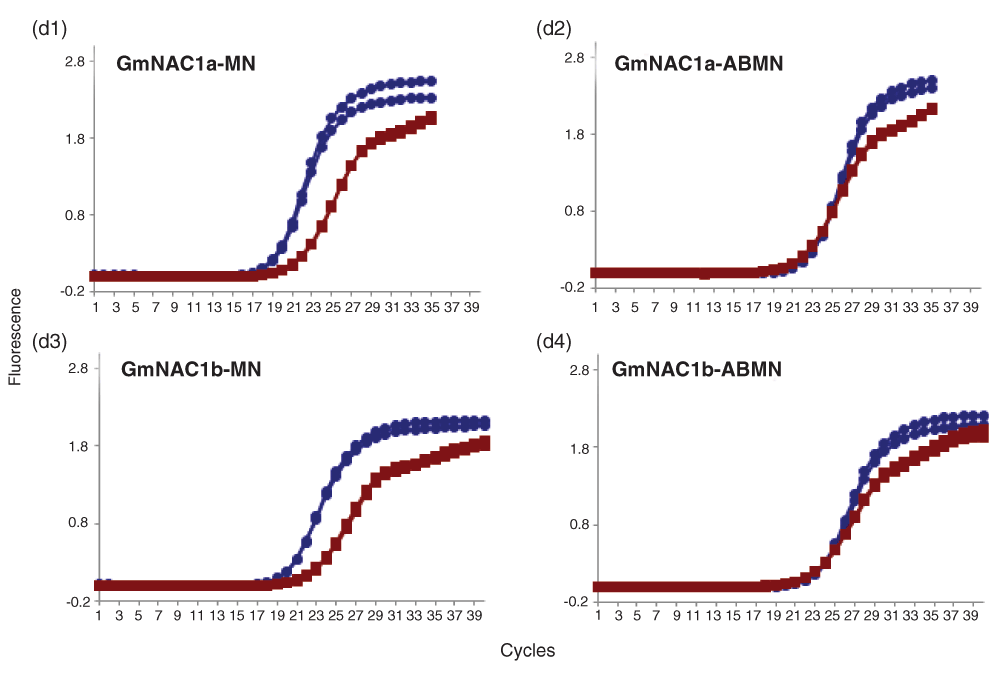
Amplification plots of GmNAC1a & b in (d1 & d3) MN (d2 & d4) ABMN tissues of soybean. Amplification plot showing linear amplification of full length (blue circles) and cleaved cDNA (red squares) by qPCR-F & R and qACE-F & R primers respectively for GmNAC1a (d1 & d2), GmNAC1b (d3 & d4) determined by SYBR fluorescence. The data shown are normalized fluorescence from two replicate assays.
| Lane 1 | Lane 2 | Lane 3 | ||
|---|---|---|---|---|
| 0hr | 24hr | 48hr | ||
| Band intensity | NAC1-Full length | 17102.98 | 8605.187 | 8547.208 |
| NAC1-Cleaved | 1067.113 | 456.062 | 760.749 | |
| Ratio(RCT) | 0.06239 | 0.05300 | 0.08901 |
Ratio of cleaved transcripts to full length in Arabidopsis plants over-expressing miR164 in response to an exogenous inducer. Previously published Northern data (Figure 4c in 33) was analyzed by quantifying the integrated band intensity using ImageJ (http://rsbweb.nih.gov/ij/). The full length and cleaved transcripts are labeled NAC1 and NAC1 3’ respectively in the original figure.
Ratio of cleaved transcripts to full length in wild-type and mir164 mutant Arabidopsis seedlings. Previously published Northern data (Figure 5c in 33) was analyzed by quantifying the integrated band intensity using ImageJ (http://rsbweb.nih.gov/ij/). The full length and cleaved transcripts are labeled NAC1 and NAC1 3’ respectively in the original figure.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)