Introduction
Our fundamental knowledge of DNA structure is based on the Watson-Crick model of DNA double helix, in which two polynucleotide chains running in opposite direction are held together by hydrogen bonds between the nitrogenous bases. Guanine can bind specifically only to cytosine (G-C) whereas adenine can bind specifically to thymine (A-T). These reactions are described as base pairing and the paired bases are said to be “complementary”1. Conformational polymorphism of DNA is now extending beyond the Watson-Crick double helix. In 1986, using forced field calculation for a short ‘A-T’ rich DNA, Pattabiraman proposed the hypothesis that homopolymeric duplex DNA containing d(A)6d.(T)6 can form a thermodynamically stable parallel right-handed duplex DNA with reverse Watson-Crick base pairing. He also reported that the number and type of hydrogen bonds between A-T base pair are the same as that of antiparallel double helix2. In 1988, the experimental strategies by Ramsing and Jovin confirmed that DNA containing A-T base pairs can exist as a stable parallel-stranded helix. The “Tm” value of both PS-DNA (parallel-stranded DNA) and APS-DNA (antiparallel-stranded DNA) showed a classical dependence upon salt concentration. They reported that at any given NaCl concentration, the melting temperature of PS-DNA was 15°C lower than its APS-DNA counterpart. In 2 mM MgCl2, the melting temperature for PS-DNA and APS-DNA was reported approximately same as those obtained in 0.2–0.3 M NaCl, demonstrating pronounced stabilization afforded by divalent cations3. A similar study by Sande et al. on hairpin deoxyoligonucleotides having oligonucleotides sequence in parallel polarities (PS-hairpin) also confirmed the existence of parallel-stranded conformation. They have shown that parallel-stranded hairpins form stable duplex and get denatured at 10°C lower than corresponding APS oligomers4. These two experimental studies provided evidence that DNA containing “A-T” base pairs can form both PS-DNA and APS-DNA. In 1992, Tchurikov et al. showed that parallel complementary probes of normal nucleotide consisting of both AT/GC base pairs can be used for molecular hybridization experiments, indicating the stability of G-C containing parallel DNA5. In 1993, Borisova et al. reported that G-C pairs in a 40 base pair parallel duplex DNA (consisting of natural DNA sequence) are more thermostable than A-T base pairs6. Furthermore, other similar reports have shown that there are no drastic differences in nearest neighbor base pair interactions between PS-DNA and APS-DNA having mixed AT/GC composition7. The specificity of the interaction between the strands in parallel DNA has also been studied and it is so high that parallel probe as short as 40 nucleotide length is able to detect a specific band in Southern blot hybridizations on whole genome DNA8. The polymerase chain reaction (PCR) developed by Mullis consists of denaturation of double-stranded DNA, primer annealing and extension. The process is repeated multiple times and the template DNA is amplified millions of times without any change in polarity of DNA9 (Figure 1). In 2000, Veitia and Ottolenghi reported that several attempts to amplify L15253 by PCR using different pairs of primers were unsuccessful. They suggested that there are no thermodynamic constraints which will prevent parallel nucleic acid synthesis, and the deoxynucleotide triphosphates used for a normal antiparallel polymerization reaction can also serve for a parallel reaction, provided that the polymerase enzyme is capable in catalyzing the nucleophilic interaction between the 3´OH and a 5´PPP from nucleotides arranged in a parallel way with respect to the template DNA10.
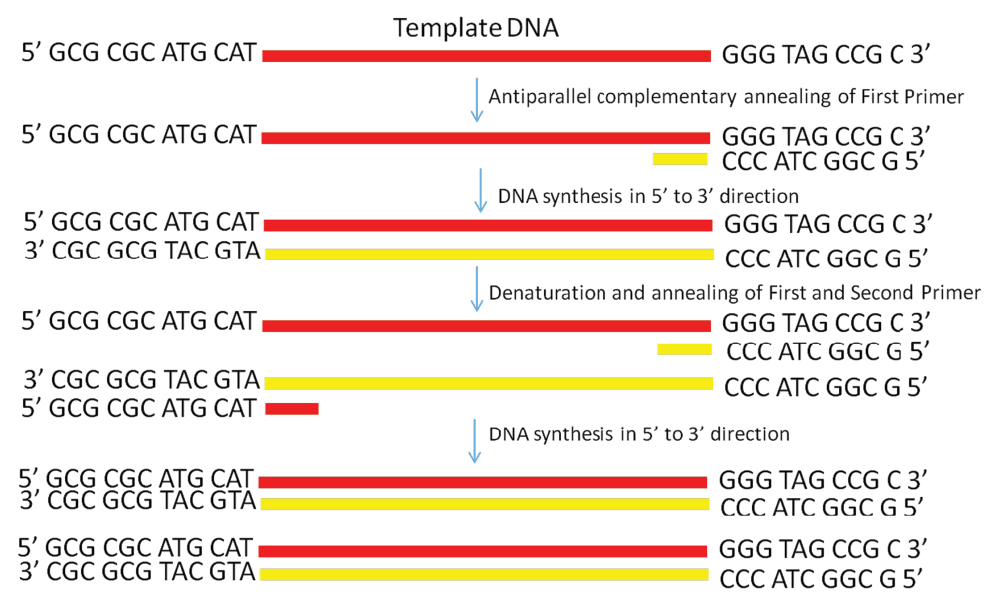
Figure 1. Schematic diagram showing PCR amplification of a single-stranded DNA by using conventional antiparallel oligonucleotide primers.
In this study, we explored whether parallel DNA synthesis is feasible. We proposed the hypothesis that this reaction can be possible if we start a reaction using single stranded DNA as a template. We have shown that the Taq DNA polymerase can even extend the oligonucleotide primer annealed to single stranded DNA in a parallel complementary manner. The details of how our proposed parallel DNA PCR differs from the conventional PCR is shown in Figure 1 and Figure 2.

Figure 2. Schematic diagram showing PCR amplification of a single-stranded DNA by using the PD-PCR (parallel DNA PCR) approach in which the first primer binds to the template DNA in a parallel complementary manner.
The second primer binds to the newly synthesized DNA in an antiparallel manner and later both primers amplify the new DNA in a conventional manner. PCR products obtained will have opposite polarity as compared to the template used.
Materials and methods
PCR
PAGE purified single stranded DNA of 120 bp was commercially obtained at a scale of 1 O.D. from Sigma Aldrich, USA. PCR oligonucleotide primers were also purchased at a scale of 0.05 O.D. from Sigma Aldrich. The sequence of custom synthesized template DNA and oligonucleotide primers used in the study are shown in Table 1. In the PD-PCR reaction, we used (PD-PCR-1) and (PD-PCR-2) primer set while for conventional PCR we used (PCR-1) and (PCR-2) primers (see Table 1). Rest of the reaction remained same. The details of PCR reaction mix were as follows: total reaction mix=50µl, primers=1µl each (50 picomole), Taq DNA polymerase=0.5µl (5U/µl), dNTP mix=0.5µl (10mM), 10X PCR buffer=5µl, water=39µl and template DNA=3µl (0.114 ng). Taq DNA polymerase (M0273S) and dNTP mix (N0447S) were purchased from NEB (New England Biolabs). PCR analysis was performed using Veriti® Thermal Cycler (Applied Biosystem) by taking single stranded template DNA and amplifying it for 30 cycles at varying annealing temperature viz. 45°C, 50°C, 55°C, 58°C, 60°C, 65°C. PCR programming included 30 cycles of denaturation at 95°C for 15 seconds, annealing at varying temperatures for 30 seconds (as explained above) and extension at 72°C for 30 seconds.
Table 1. Shows sequence of custom synthesized template DNA and oligonucleotide primers used in this study.
Sequence of single stranded
template DNA | 5´ GCG CGC ATG CAT GTG ACT GAC GAT CGA TCG ATC AGT ACT GAC
TGA CAA ATG ACT GGA TCC GGG AAG CTT GTG TTT AAA GTG TGA
GGG TTG GCT GGG GTG TGG GG G TG GAT GGG TAG CCG C 3´ |
Oligonucleotides primers for
conventional PCR (Figure 1) | (PCR-1)
5´GCG GCT ACC CAT CCA CCC CCA C 3´
(PCR-2)
5´ GCG CGC ATG CAT GTG ACT GAC 3´ |
Oligonucleotides primers for
PD-PCR (Figure 2) | (PD-PCR-1)
5´CGC GCG TAC GTA CAC TGA CTG C 3´
(PD-PCR-2)
5´ CGC CGA TGG GTA GGT GG GGG 3´ |
Agarose gel electrophoresis
PCR products obtained in two reactions were separated on 1% agarose gel containing ethidium bromide and were run and observed in gel-doc (DNR Bioimaging system, Jerusalem, Israel) under UV. Electrophoresis apparatus used in these experiments was purchased from Chromus Biotech, Bengaluru, India whereas chemicals {Agarose (A9539) and EtBr (E7637)} were purchased from Sigma, USA.
Sequencing
The amplified products were sequenced at Eurofins Genomics India Pvt. Ltd. Karnataka India.
Real time PCR
Real time PCR was performed with 2X SYBR Green master mix (K0221, Thermo Scientific, Pittsburgh, USA). The details of real time PCR reaction mix were as follows: total reaction mix=10µl, primers=0.25µl each, (50 picomole), 2X SYBR green mastermix=5µl, water=4µl and template=0.5µl (1:1000 dilution from original stock of 0.96 picomole). In the PD-PCR reaction, we used (PD-PCR-1) and (PD-PCR-2) primer set while for conventional PCR we used (PCR-1) and (PCR-2) primers (see Table 1). Negative control included reaction mix without template DNA. Reactions were incubated at 94°C for 5 minutes, followed by 30 PCR cycles of 94°C for 15 seconds, 50°C for 30 seconds and 72°C for 60 seconds using Mx3005P qPCR System - Agilent Technologies, Inc. The data were analyzed by using 2−ΔCt method. The products were also run on agarose gel and visualised on gel doc system as previously described.
Result and discussion
Dataset 1.DNA sequencing file for PCR and PD-PCR.
DNA sequencing file (DNA sequencing file for PCR.ab1) confirming single stranded template DNA was amplified as it is while DNA sequencing file (DNA sequencing file for PD-PCR.ab1) confirming that single stranded template DNA was amplified as per our proposed PD-PCR scheme within this manuscript.Dataset 2.Real time PCR file for PCR and PD-PCR.
Real time PCR amplification of single stranded DNA as per conventional PCR and PD-PCR.1 Data fileThe thermal denaturation analysis of parallel DNA has shown that it melts at a lower temperature than the corresponding antiparallel structure3,4. This finding gives us the clue that using double-stranded antiparallel DNA as a template for PD-PCR will not be possible as during annealing steps, antiparallel double-stranded DNA will anneal to itself without binding to parallel-stranded complementary primers. To avoid this, we started our PCR with a single-stranded DNA template. Details on how our proposed parallel DNA PCR (PD-PCR) differs from conventional PCR are shown in Figure 2. The first oligonucleotide primer (PD-PCR-1) was designed to bind the single-stranded template DNA in a parallel complementary manner. The parallel complementary annealing of the first primer allowed the synthesis of DNA in a parallel direction to the single-stranded DNA template. After the first denaturation step, the second oligonucleotide primer (PD-PCR-2) was designed to anneal to the newly synthesized DNA in an antiparallel complementary orientation. Further, both first and second primers used in this reaction amplified the new second DNA strand in a conventional way by binding in an antiparallel complementary way. Figure 3 (lanes 8–13) shows a 120 bp PCR product amplified by parallel DNA PCR scheme at annealing temperature of 45°C, 50°C, 55°C, 58°C, 60°C, 65°C respectively. In all cases, denaturation was performed at 95°C for 15 seconds, annealing for 30 seconds while extension at 72°C for 30 second for a total of 30 cycles. Similarly, as a control reaction, the single-stranded 120 bp DNA was amplified by conventional PCR in which the first primer (PCR-1) bound to the template DNA in an antiparallel orientation and the second primer (PCR-2) annealed to the newly synthesized DNA in an antiparallel orientation. Figure 3, Lanes 1–6 shows a 120 bp product PCR amplified at annealing temperature of 45°C, 50°C, 55°C, 58°C, 60°C, 65°C respectively using conventional antiparallel complementary primers. As a control reaction, PD-PCR was also performed using only one of the two primers. As expected, no PCR products were obtained (Figure 4A lane 2 and 3). As a control reaction, conventional PCR and PD-PCR were performed without adding any template DNA. As expected, no PCR product was obtained confirming that no primer dimer was formed during both conventional PCR and PD-PCR (Figure 4B). The DNA sequencing results confirmed that DNA templates were amplified in two different PCR products. Conventional PCR amplified the template DNA in its original orientation (Figure 5A) whereas PD-PCR products read in a parallel direction to the template DNA (Figure 5B). Primers and template used to show feasibility of PD-PCR till now (Figure 3 and Table 1) were further used to perform real-time PCR. For this, a master mix containing SYBR Green and other components (except template and primers) was used. Primers and template were added to the master mix to make a final volume of 10µl. A Ct value of 9.26 was obtained for conventional PCR, 23.29 for PD-PCR whereas 33.15 was observed in negative control (without adding template DNA) indicating amplification in both conventional PCR and PD-PCR reactions (Figure 6). The amplification indicated by Ct value in real time PCR was also confirmed by running the product on agarose gel (Figure 6, lower panel). Weak amplifications in PD-PCR may be attributed to the fact that the actual amplification in conventional PCR is one step ahead than the PD-PCR. The first amplification in conventional PCR starts as early as denaturation followed by annealing (Figure 1). On the other hand, in PD-PCR a new template is first synthesized during the first amplification (represented by green color in Figure 2). Once the template is ready, the conventional PCR goes on. Therefore, the amplified product shows low intensity as compared to the conventional PCR products. Taking together, our study has shown that DNA synthesis can happen in a parallel direction and two different, but related PCR products can be synthesized from the single-stranded template DNA. We hope that more molecular biology techniques will develop in future based on parallel complementary bindings of duplex DNA.
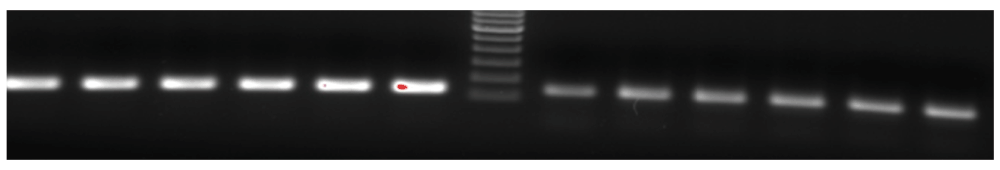
Figure 3. PD-PCR (parallel DNA PCR) and PCR: Lanes 1–6 show 120 bp PCR products amplified at annealing temperature of 45°C, 50°C, 55°C, 58°C, 60°C, 65°C, respectively, using conventional antiparallel complementary primers.
Lane 7 is 100 bp molecular weight marker and Lanes 8–13 show PCR products amplified by parallel DNA PCR scheme at annealing temperature of 45°C, 50°C, 55°C, 58°C, 60°C, 65°C, respectively. In all cases, denaturation was performed at 95°C for 15 seconds, annealing for 30 seconds while extension at 72°C for 30 second for a total of 30 cycles.
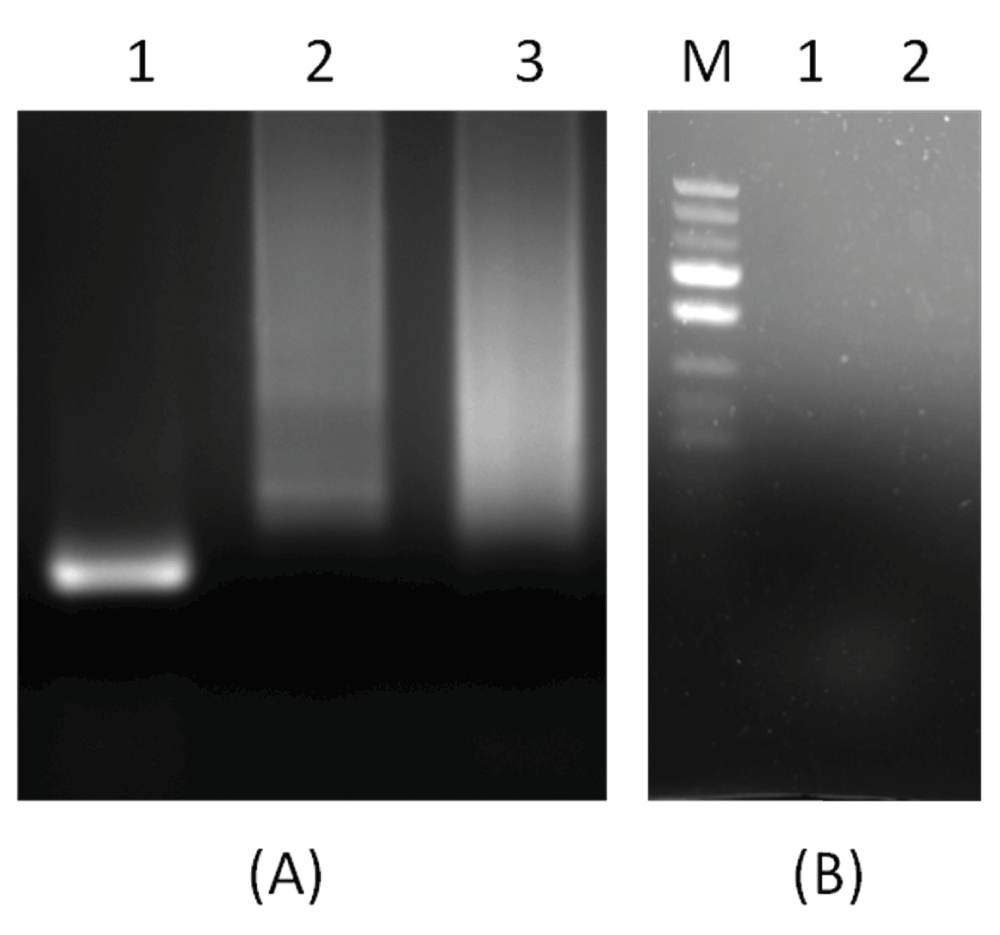
Figure 4.
(A): A control reaction showing that PCR products were obtained when both primers were added as per scheme in Figure 2. In Figure 4 (A), Lane 1 shows 120 bp PCR products synthesized by PD-PCR, while in Lanes 2 and 3, only single primers were added and as expected no PCR product was synthesized. Figure 4(B) shows a negative control reaction of conventional PCR and PD-PCR in which the template DNA was not added.
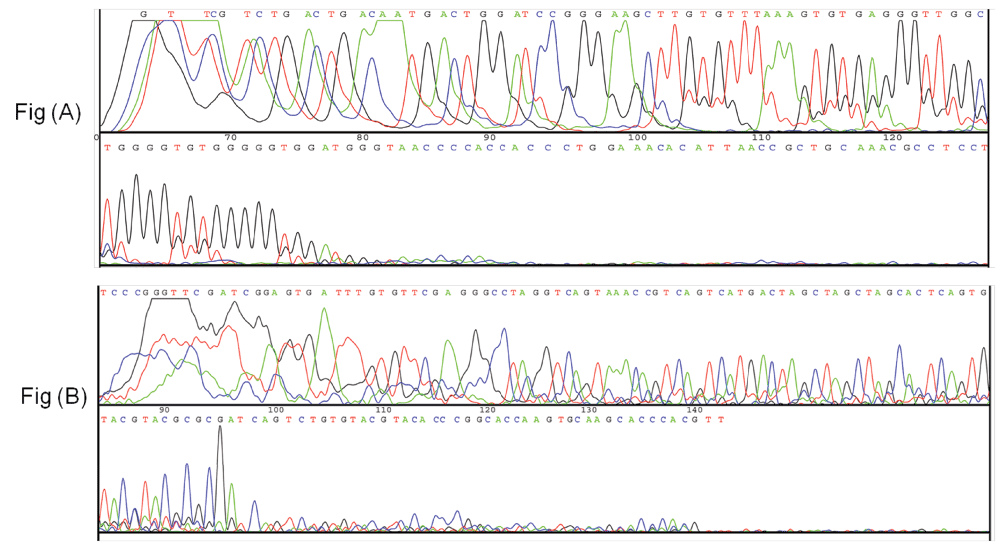
Figure 5.
DNA sequencing results. Sequencing results in (A) show that 120 bp DNA was amplified as it is while sequencing results in (B) confirm that PCR products were obtained as per the scheme shown in Figure 2.
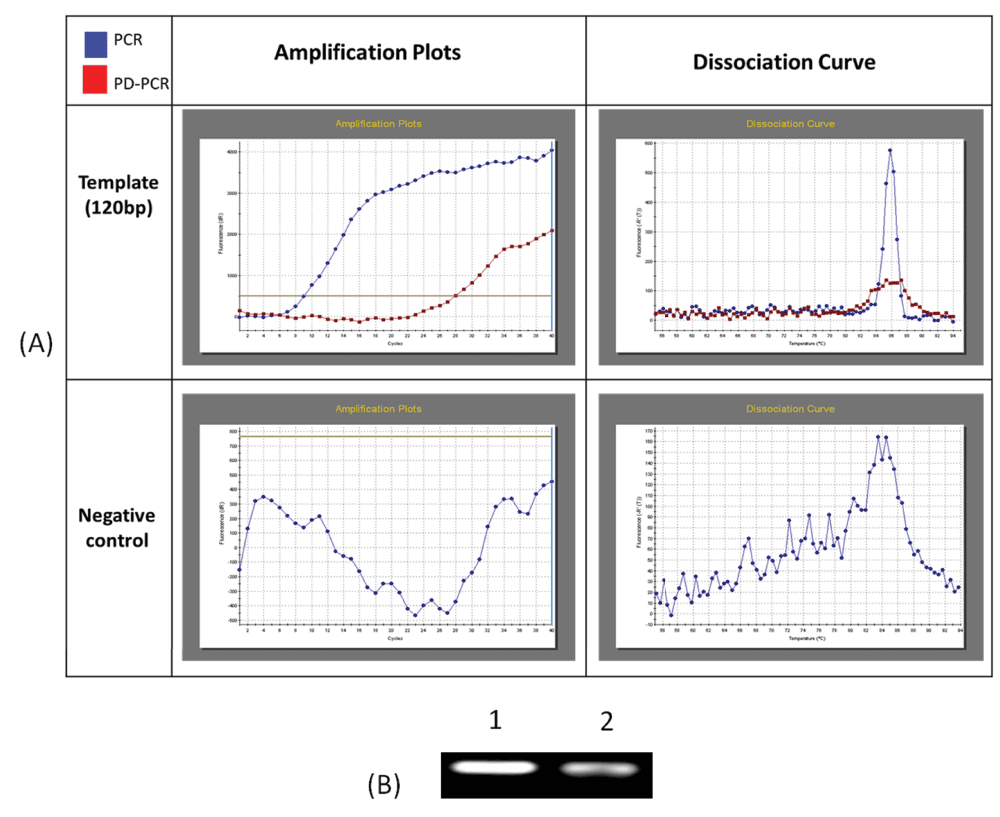
Figure 6.
Real time PCR and PD-PCR (A) show ampliflication plot and dissociation curves obtained after real time PCR analysis of amplification of 120 nucleotides single stranded template DNA via conventional PCR and PD-PCR. In control reaction no template DNA was added. (B) PCR products obtained in real time PCR were also run on agarose gel and visualised on gel doc system.
Data availability
F1000Research: Dataset 1. DNA sequencing file for PCR and PD-PCR. 10.5256/f1000research.5813.d4151511
F1000Research: Dataset 2. Real time PCR file for PCR and PD-PCR. 10.5256/f1000research.5813.d4151612
Author contributions
VB and KS designed the experiment. VB and KS carried out the research. Both prepared the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Acknowledgments
We are thankful to Harpreet Singh (Territory Manager, Sigma-Aldrich, Gujarat, India) for providing the custom synthesized template DNA and oligonucleotide primers used in this study.
Faculty Opinions recommendedReferences
- 1.
Watson JD, Crick FH:
Molecular structure of nucleic acids; a structure for Deoxyribose Nucleic Acid.
Nature.
1953; 171(4356): 737–738. PubMed Abstract
| Publisher Full Text
- 2.
Pattabiraman N:
Can the Double Helix Be Parallel?
Biopolymers.
1986; 25(9): 1603–1606. PubMed Abstract
| Publisher Full Text
- 3.
Ramsing NB, Jovin TM:
Parallel stranded duplex DNA.
Nucleic Acids Res.
1988; 16(14A): 6659–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
van de Sande JH, Ramsing NB, Germann MW, et al.:
Parallel Stranded DNA.
Science.
1988; 241(4865): 551–557. PubMed Abstract
| Publisher Full Text
- 5.
Tchurikov NA, Shchyolkina AK, Borissova OF, et al.:
Southern molecular hybridization experiments with parallel complementary DNA probes.
FEBS Lett.
1992; 297(3): 233–236. PubMed Abstract
| Publisher Full Text
- 6.
Borisova OF, Shchyolkina AK, Chernov BK, et al.:
Relative stability of AT and GC pairs in parallel DNA duplex formed by a natural sequence.
FEBS Lett.
1993; 322(3): 304–6. PubMed Abstract
| Publisher Full Text
- 7.
Shchyolkina AK, Borisova OF, Livshits MA, et al.:
[Parallel-stranded DNA with natural base sequences].
Mol Biol.
2003; 37(2): 223–231. PubMed Abstract
| Publisher Full Text
- 8.
Tchurikov NA, Shchyolkina AK, Borissova OF, et al.:
Southern molecular hybridization experiments with parallel complementary DNA probes.
FEBS Lett.
1992; 10: 297(3): 233–6. PubMed Abstract
| Publisher Full Text
- 9.
Mullis KB:
Process for amplifying nucleic acid sequences, United States Patent 4683202. 1987. Reference Source
- 10.
Veitia R, Ottolenghi C:
Placing parallel stranded DNA in an evolutionary context.
J Theor Biol.
2000; 206(2): 317–322. PubMed Abstract
| Publisher Full Text
- 11.
Bhardwaj V, Sharma K:
Dataset 1. DNA sequencing file for PCR and PD-PCR.
F1000Research.
2014. Data Source
- 12.
Bhardwaj V, Sharma K:
Dataset 2. Real time PCR file for PCR and PD-PCR.
F1000Research.
2014. Data Source
{kind=link}
I made some experiments on to test a possibility of parallel synthesis in March, 2015. I used the template corresponding to that described in your paper, but shorter:
5’GCGCGCATGCATGTGACTGACGATCGATCGATCAGTACTGACTGACAAATGAGTGGGGGTGGATGGGTAGCCGC3’(template, ... Continue reading Dr. Vikash Bhardwaj,
I made some experiments on to test a possibility of parallel synthesis in March, 2015. I used the template corresponding to that described in your paper, but shorter:
5’GCGCGCATGCATGTGACTGACGATCGATCGATCAGTACTGACTGACAAATGAGTGGGGGTGGATGGGTAGCCGC3’(template, Olig №1).
In bold the region located between the primers and designated for testing of parallel synthesis is shown.
Expected par-template (see below)could appear after extension of a primer [par(+) 5’CGCGCGTACGTACACTGACTGC 3’] that is complementary in parallel orientation to the template. It was performed using a mixture of several DNA polymerases including three thermostable ones - at 37°C and then at 72°C.
The sequence of expected DNA is:
5’CGCGCGTACGTACACTGACTGCTAGCTAGCTAGTCATGACTGACTGTTTACTCACCCCCACCTACCCATCGGCG3’ (par-template).
For amplifications of putative par-template I used the mixture after this primer extension, containing the chemically synthesized template (in great molar excess!!!) and, as I suggest cannot be excluded, traces of putative par-template, that may correspond to not complete sequence of the shown par-template.
For regular PCR amplification of par-template two primers were used - №4- par(+)5’CGCGCGTACGTACACTGACTGC 3’and №5- par(-) 5’ CGCCGATGGGTAGGTGGGGG 3’, that are complementary to par-template in antiparallel orientation.
The same mixture were used for amplification of template (№1) and primers №2 ap(+) 5’GCGCGCATGCATGTGACTGACG 3’ and №3 ap(-) 5’GCGGCTACCCATCCACCCCCAC 3’.
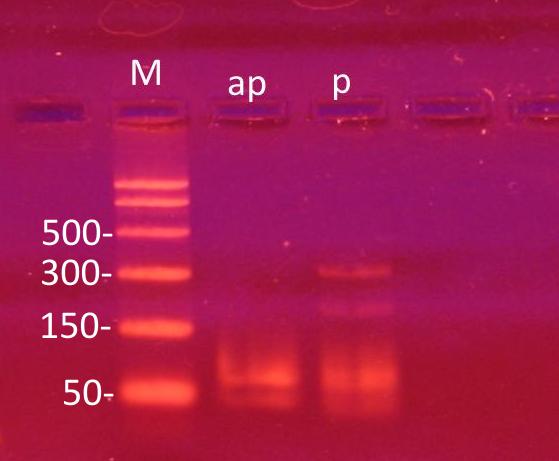
The results are shown in the linked file. ap - PCR with primers №1 and 2; p - PCR with primers №4 and №5.
The amplified DNAs were cloned and sequenced. All ap-clones corresponded to the oligos №1, as expected.
About 50 par-clones were sequenced. Mostly they correspond to the text of original template shown in bold (in oligos №1) located between par-primers, indicating that primers №4 and №5 are capable to anneal to the original template that is present in a great excess.
Two par-clones (#11 and 42) were different and possibly possess some short sequences that could be explained either by extension of primer № 4 in parallel orientation, or by combination of sequences of original olig №1 and primer №5. I have no time for a careful analysis. E.g. - GCGGCTACCCATCCACCCCCACCTACCCATCGGCG 3’- mirror hairpin with a loop in #42.
I am sorry that this nice gel gave such clones.
New experiments with well thought template sequences (may be with low GC-content) could be used. Only sequence of individual clones should be performed. The careful analysis of unusual short stretches of mirror sequences should be performed. I suggest to reduce dramatically the content of original template using 5'-biotinylated templates that could be separated from par-template using PMP-particles.
Sincerely, Nickolai.
P.S. I should say that the mirror sequences in these experiments may also mean simply annealing of (-) primer designed for regular amplification of par-template in the 3' region of original template which possess mirror stretch of the same nucleotides.
I made some experiments on to test a possibility of parallel synthesis in March, 2015. I used the template corresponding to that described in your paper, but shorter:
5’GCGCGCATGCATGTGACTGACGATCGATCGATCAGTACTGACTGACAAATGAGTGGGGGTGGATGGGTAGCCGC3’(template, Olig №1).
In bold the region located between the primers and designated for testing of parallel synthesis is shown.
Expected par-template (see below)could appear after extension of a primer [par(+) 5’CGCGCGTACGTACACTGACTGC 3’] that is complementary in parallel orientation to the template. It was performed using a mixture of several DNA polymerases including three thermostable ones - at 37°C and then at 72°C.
The sequence of expected DNA is:
5’CGCGCGTACGTACACTGACTGCTAGCTAGCTAGTCATGACTGACTGTTTACTCACCCCCACCTACCCATCGGCG3’ (par-template).
For amplifications of putative par-template I used the mixture after this primer extension, containing the chemically synthesized template (in great molar excess!!!) and, as I suggest cannot be excluded, traces of putative par-template, that may correspond to not complete sequence of the shown par-template.
For regular PCR amplification of par-template two primers were used - №4- par(+)5’CGCGCGTACGTACACTGACTGC 3’and №5- par(-) 5’ CGCCGATGGGTAGGTGGGGG 3’, that are complementary to par-template in antiparallel orientation.
The same mixture were used for amplification of template (№1) and primers №2 ap(+) 5’GCGCGCATGCATGTGACTGACG 3’ and №3 ap(-) 5’GCGGCTACCCATCCACCCCCAC 3’.
The results are shown in the linked file. ap - PCR with primers №1 and 2; p - PCR with primers №4 and №5.
The amplified DNAs were cloned and sequenced. All ap-clones corresponded to the oligos №1, as expected.
About 50 par-clones were sequenced. Mostly they correspond to the text of original template shown in bold (in oligos №1) located between par-primers, indicating that primers №4 and №5 are capable to anneal to the original template that is present in a great excess.
Two par-clones (#11 and 42) were different and possibly possess some short sequences that could be explained either by extension of primer № 4 in parallel orientation, or by combination of sequences of original olig №1 and primer №5. I have no time for a careful analysis. E.g. - GCGGCTACCCATCCACCCCCACCTACCCATCGGCG 3’- mirror hairpin with a loop in #42.
I am sorry that this nice gel gave such clones.
New experiments with well thought template sequences (may be with low GC-content) could be used. Only sequence of individual clones should be performed. The careful analysis of unusual short stretches of mirror sequences should be performed. I suggest to reduce dramatically the content of original template using 5'-biotinylated templates that could be separated from par-template using PMP-particles.
Sincerely, Nickolai.
P.S. I should say that the mirror sequences in these experiments may also mean simply annealing of (-) primer designed for regular amplification of par-template in the 3' region of original template which possess mirror stretch of the same nucleotides.