Keywords
Ehlers-Danlos syndrome, arthrochalasia, orthopaedic surgery, treatment
Ehlers-Danlos syndrome, arthrochalasia, orthopaedic surgery, treatment
Ehlers-Danlos syndrome (EDS) is a well known inherited connective-tissue disorder characterized by general joint hypermobility, tissue fragility and skin abnormalities. Six subtypes are described which show clinical overlap with each other and other syndromal connective tissue disorders (e.g. Larsen syndrome)1,2. EDS arthrochalasia type (former EDS type VII-A and VII-B) can be distinguished from other types by the presence of mild dysmorphic features (in particular hypertelorism and micrognathia), congenital bilateral hip dislocation, multiple other recurrent (sub)dislocations and severe muscular hypotonia. This type of EDS is very rare with only 27 cases described to-date1.
In this case report the history of a three year old girl with EDS arthrochalasia type is described. This girl has been described previously1.
A 6 year old Caucasian girl, known in the paediatric, clinical genetic and orthopaedic outpatient clinic since birth, was treated for severe skin problems, hyperlaxity of her joints and hypotonia. These congenital symptoms were initially thought to be a part of Larsen syndrome, because of the facial features and hypotonia. However, this would not explain the severe skin problems. At the first visit to our outpatient clinic, when the girl was four months, a bilateral hip dislocation in 90° abduction was seen. Manual closed reduction was considered a possibility. The shoulders dislocated spontaneously with maximum anteversion. X-rays showed high dislocations of the both hip joints and a thoracolumbar scoliosis (Figure 1 and Figure 2). At the age of six months EDS arthrochalasia type was suspected and confirmed by Sanger sequencing, which showed a COL1A2 mutation.
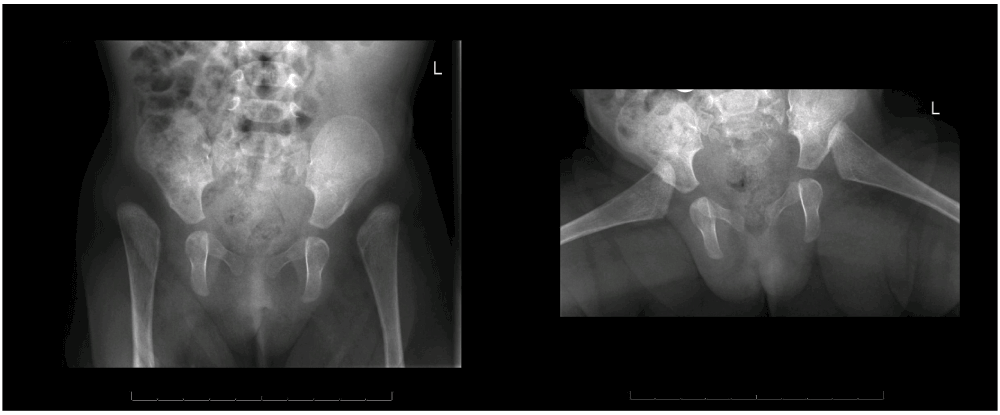
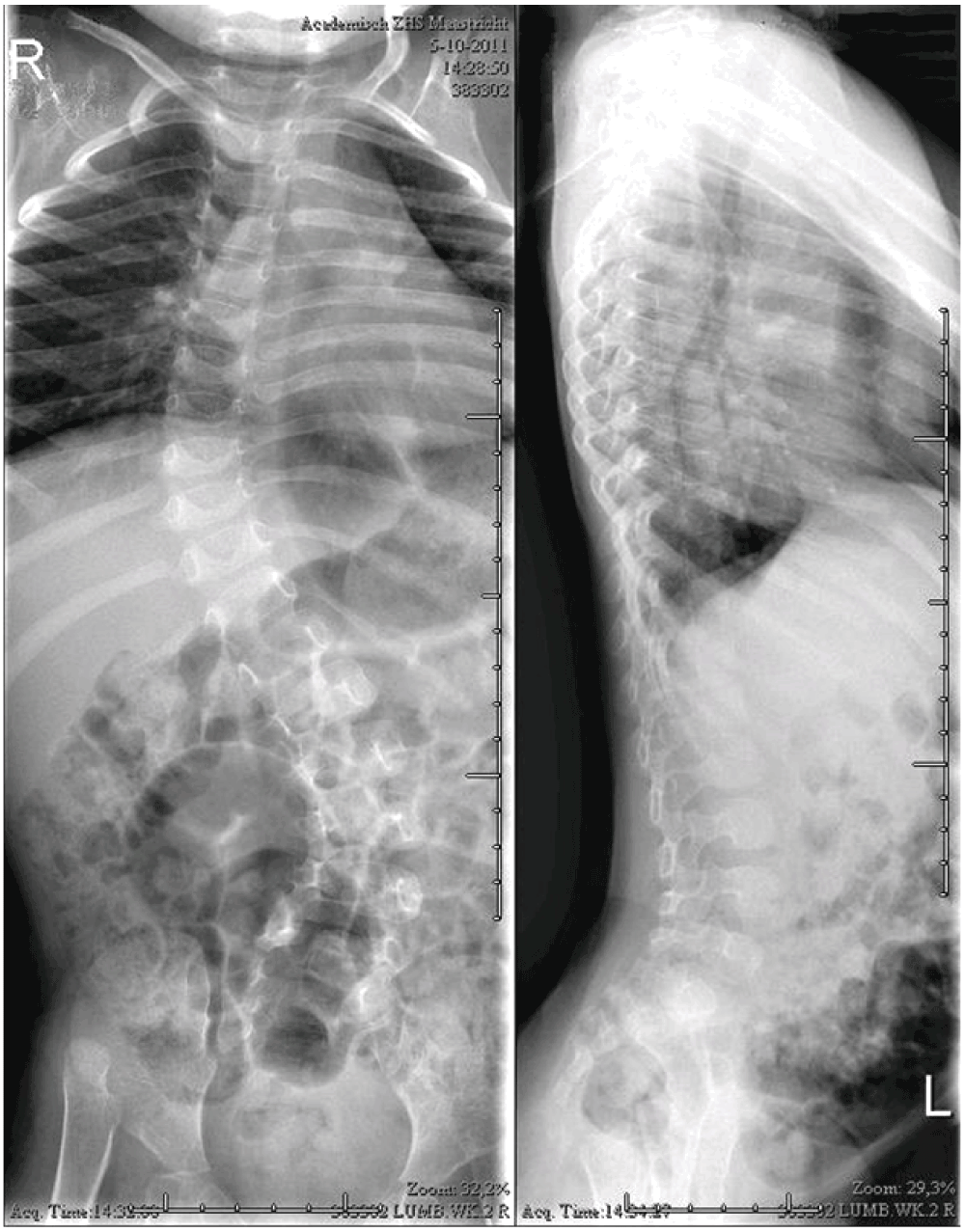
Initially, the hip dislocation was not treated with a harness because of the easy bruising and multiple lacerations of the skin, and the unknown origin of the syndrome. After confirmation of the diagnosis, surgical hip reduction in the near future was believed not to be the best intervention. The reason for this was the high chance that obtaining a stable reposition would fail, because of the severe connective tissue abnormalities. Given the severe hypotonia and multiple other dislocations, the prognosis for walking was small. In addition, the patient would have to wear a plaster cast for at least 12 weeks postoperatively, which would cause major skin problems and would probably further impair her motor development. For the same reason conservative treatment in terms of only a plaster cast was deemed unachievable. This decision was supported by findings in the literature showing that conservative treatment in these cases is not successful.
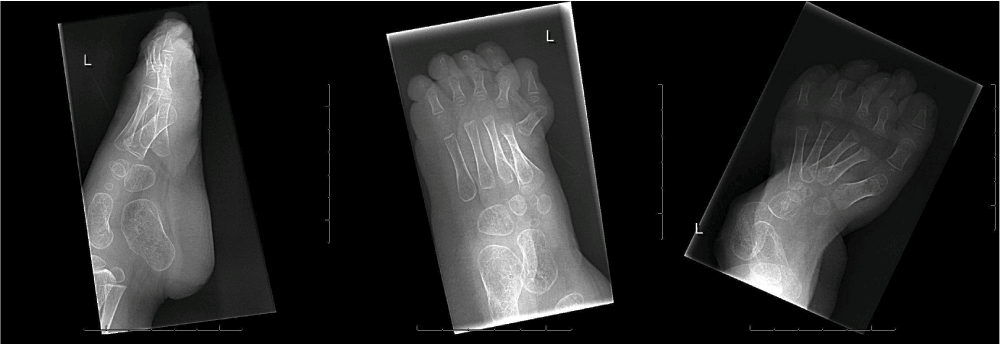
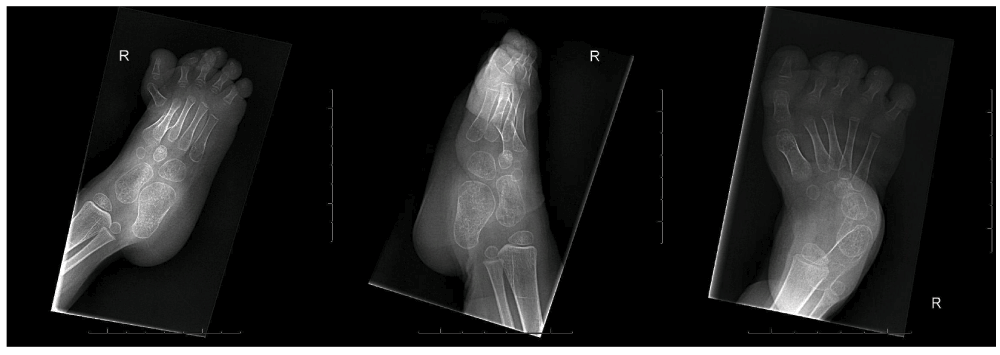
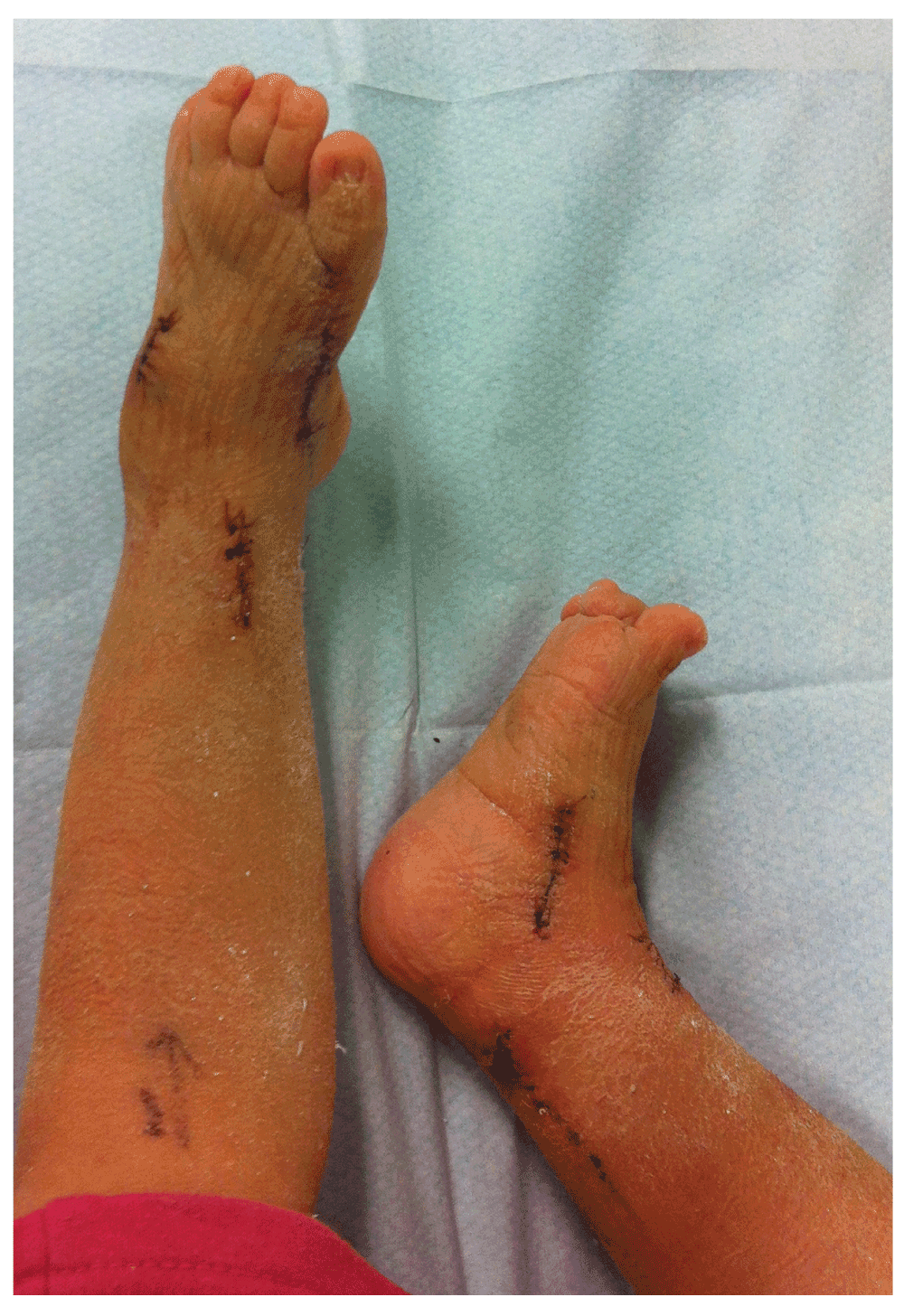
Follow-up of the patient showed a girl with a progressive equinovarus deformity of both feet (Figure 3–Figure 5) starting at the age of 1 year and 6 months. Due to her foot deformity, bilateral hip dislocation and hypotonia it was not possible for the girl to mobilize. Initially, the foot deformity was mild and easy to redress. After one year (at age 2 years and 6 months) it was no longer possible to redress the foot deformity, due to contractures and stiffness of the joints. A decision for surgical treatment was made in order to allow shoe wearing. A bilateral tenotomy of the Achilles tendon and a bilateral anticus transfer was performed. Extension of the flexor hallucis tendon of the right foot and a tenotomy of the flexor hallucis tendon of the left foot were also part of the treatment. Postoperative treatment consisted of a well-padded plaster cast for 6 weeks and appropriate footwear afterwards. After this treatment, a slight foot deformity remained (Figure 6), although the intention of treatment, which was to redress the deformity in her feet so that they would fit shoes, was achieved.

In 1892, Tschernogobow was the first to describe joint dislocations due to ligamentous and capsular laxity4. Ehlers-Danlos syndrome is an inherited connective-tissue disorder with an autosomal dominant mode of inheritance3. General clinical symptoms are hypermobility of the joints, tissue laxity and skin abnormalities. There are six different subtypes, based on their clinical and genetic features. EDS arthrochalasia type is caused by mutations in the COL1A1 (OMIM 130060) or COL1A2 (OMIM 130060) gene, which causes production of low quality type 1 collagen fibers. This EDS type distinguishes itself from the other types of EDS by the severity of the congenital bilateral hip dislocations, recurrent subluxations, subtle dysmorphic features, a severe muscular hypotonia, a soft velvety skin that is not hyperextensible and a ‘criss-cross’ patterning of the palms and soles1,3.
Of all forms of EDS, approximately 90% are the classic and hypermobile subtypes (EDS I & III) and 5–10% is the vascular subtype (IDS IV). All the other subtypes are extremely rare. Only 27 cases of EDS arthrochalasia type are described in the literature1,2.
A postnatal early diagnosis of EDS arthrochalasia type is important, because an early diagnosis will benefit follow-up and treatment. Important issues compromising development, such as the severe hypermobility with dislocations and hypotonia, can be addressed appropriately. Establishing a diagnosis can be difficult in the neonatal period, because of phenotypic overlap with other skeletal dysplasias, such as Larsen syndrome1,3.
There is no consensus in literature on treatment of EDS arthrochalasia type. The little knowledge there is on treatment of the upper limbs tells us that neither orthotics nor surgical treatment are effective5.
With regard to the lower limbs, a choice between conservative treatment vs. surgical treatment is an important step in the treatment process. Conservative treatment, such as plaster casts, Pavlic bandages or orthotics, would be preferable, but they have a high chance of causing decubitus lesions, due to high pressures on a highly vulnerable skin. Therefore, these treatments are not favoured in patients with motor retardation. Also for lower limbs it seems, according to the literature, that conservative treatment is not effective. Surgical treatment is effective in only some cases in the treatment of congenital hip dislocations (Table 1)5. Only an iliac osteotomy with or without a varus and derotation femoral osteotomy shows stability in other hyperlaxity syndromes1,5.
For the treatment of equinovarus deformities of the feet, which are common in EDS arthrochalasia type patients, treatment options are also described to be rarely effective6.
Although surgical procedures are invasive, they seem to be the only proven treatment to be effective. In this case a bilateral hip dislocation and an equinovarus deformity of both feet was seen and conservative treatment was considered not to be effective or to possibly be even harmful in this child. Therefore, a capsular and tendon release of the feet was carried out. Another treatment option for the feet could have been a subtalar release or a triple arthrodesis6,7.
It is advisable to wait till the end of childhood for correction of the scoliosis deformity. In general, muscles will have gained strength during childhood, partly because of improvement in muscle tone. This most likely will result in an improvement of the scoliosis6.
EDS arthrochalasia type is rare. In all patients the dislocation of the joints combined with hypotonia is a serious problem, leading to severe disability. All interventions, both conservative and surgical, should be considered carefully. As with any treatment method, complications such as wound healing problems and decubitus, resulting from the poor quality of the collagen fibers, are a risk. The best treatment depends on prognosis and current situation of the child.
Written informed consent for publication of her clinical images was obtained from the parent of the patient.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)