Introduction
Amyotrophic lateral sclerosis (ALS) is the most feared, frequent, flummoxing and fatal motor neuron disease1–3. It is a biphasic disease which starts insidiously, later followed by relentless progression once symptomatic4. Although rare, it is a grim and demeaning illness: it slowly cripples and confines its victims in their own body, ultimately killing them by breathing failure within 3–5 years after onset2,5. No cure exists1. Ever since Charcot’s description of ALS (1869), a classical view defines ALS as an adult-onset neurodegenerative disease of upper and lower motor neurons6,7. However, ALS is clinically characterised by variability about the type and degree of motor neuron and non-motor neuron involvement8.
ALS pathology involves an interaction of multiple genes and environmental factors. Indeed, ALS is mainly a polygenic disease (70%–90); and the heritable form (familial ALS) contributes to merely 30% of total ALS cases3,9. Remarkably, mutant C9ORF72, TARDBP, FUS, and SOD1 genes account for 70% of all familial ALS cases10. Evidence shows that environmental factors such as intense physical activity, cigarette smoking, viral infections, and the ingestion of non-protein amino acids (i.e. β-N-methylamino-L-alanine) play a role in ALS5,11,12.
ALS aetiology: an enduring enigma
Despite nearly 150 years of research, ALS remains an enigma13, although, at the cellular, molecular and metabolic levels, a staggering and ever expanding list of pathogenic mechanisms have been linked to ALS13,14. These include protein aggregation, mitochondrial dysfunction, oxidative/nitrosative stress, endoplasmic reticulum (ER) stress, axonal transport defects, glutamate excitotoxicity, impaired macroautophagy, impaired glycolysis, neuroinflammation, and glucose and fat metabolism impairments13–19. However, hitherto no hypothesis exist that effectively links all these mechanisms to a singular central cause3. Hence, despite steadily accumulating knowledge about ALS, a key question still lingers: what cause ALS?
ALS: a multi organ disease
Since ALS is manifestly a neurological disorder, researchers have long embraced an intuitive neurocentric view of ALS, assuming that intrinsic neuronal pathology causes ALS7,20. Against this view, however, growing evidence suggests that ALS pathology extends well beyond neuronal cells and involves multiple organs7,14,21. Unsurprisingly, ALS is now deemed as a systems disease20,22. Not only that, evidence increasingly shows that primary pathological events, inherited or acquired, within these organs may act as distal cause of ALS14,21,22. Such evidence is reviewed below.
Role of skeletal muscle
ALS starts and spreads from skeletal muscle14,23. Indeed, some early symptoms of ALS involve the neuromuscular system: muscle atrophy, cachexia (wasting), weakness, and fasciculation (twitches)7,11,14,19,23. In fact, cachexia reduces survival of ALS patients19. Reinforcing such observations, data from animal models of ALS showed neuromuscular dysfunction precede motor neurons loss14. For instance, Frey et al. showed selective loss of fast-fatigable neuromuscular synapses of SOD1G93A mice by 6 weeks of age, 2 month before symptomatic phase24. Cogently, a study showed that the expression of mutant gene (SOD1G93A) exclusively in skeletal muscle of transgenic mice caused cachexia, neuromuscular denervation, paresis, and motor neuron degeneration25.
Skeletal muscle possesses mainly two types of muscle fibres: fast twitch and slow twitch26. Lately, evidence suggests that in ALS fast twitch muscle motor units are selectively damaged before overt symptoms, whereas slow twitch motor units show damage after overt symptoms27,28. For example, a set of studies showed a rapid motor unit loss during the presymptomatic phase (5 weeks of age) in fast but not slow-twitch muscles of the SOD1G93A mouse27. Accordingly, fast twitch muscle appears to be more susceptible to damage in ALS patients28. Therefore, fast twitch muscle pathology appears to be the distal cause of ALS.
Together, those findings have led to the “dying-back” hypothesis14. This holds that ALS is a distal axonopathy in which pathological changes first arise distally at the neuromuscular junction and progress backward toward the spinal cord cell body14. That said, however, recent and prior research mandates refinement of this hypothesis. Recently, experiments in the SOD1G93A mice showed independent and parallel degeneration of both upper and lower motor neurons at early stage, hinting at a common pathological mechanism29. Consistent with this, recent neuroimaging studies showed early stage involvement of upper motor neuron (UMN) in ALS patients30. In fact, Gower (1886), Charcot’s contemporary, suggested simultaneous and independent degeneration of upper and lower motor neurons in ALS31. Thus, a common but hitherto unidentified pathological factor emanating from skeletal muscle appears to damages both upper and lower motor neurons.
Liver: an emerging locus of ALS
Aside from skeletal muscle, mounting evidence suggests that liver dysfunction commonly occurs in ALS. Indeed, literature on the liver pathology in ALS has existed for over a half century32,33. Earlier, researchers showed a range of liver abnormalities in ALS patients including the disturbance of unconjugated bilirubin metabolism, mitochondrial defects, and copper accumulation in hepatic lysosomes32. More recently, clinical studies suggest that hepatic steatosis (fatty liver degeneration) is a common and unique phenomenon in motor neuron diseases including ALS22,34,35. Nodera et al. found that hepatic steatosis was present in 76% of ALS patients22. In line with this, studies showed reduced growth hormone/insulin-like growth factor-1 (GH/IGF-I) levels, which induce hepatic steatosis, in ALS21,36. In keeping with this, hyperhomocysteinemia, which is associated with hepatic fat accumulation, commonly occurs in ALS patients37,38. Moreover, research showed that ALS-associated environmental factors such as virus infection (i.e. retrovirus virus and HIV) and cigarette smoking cause hepatic steatosis5,12,39,40. Furthermore, viral hepatitis, which causes hepatic insufficiency and frequent fatty liver degeneration, has been linked to motor neuron disease41–44. Finally, Reye-like syndrome, associated with fatty liver degeneration, has been associated with spinal muscular atrophy (SMA), a lower motor neuron disease35.
A number of genetic findings also support this notion. Iron dysregulation disorders such as HFE gene-related hemochromatosis and hyperferritinemia, which induces hepatic steatosis, frequently (30%) occurs in ALS45,46. Additionally, mutant cholesterol and lipid pathways genes such as TDP-43 ATXN2, paraoxonase and CYP7A1, implicated in hepatic steatosis, have been linked to ALS47–55. Moreover, an interaction between disturbances in hepatic mitochondrial function and ER homeostasis causes hepatic steatosis; and investigators discovered morphological changes in ER structure and mitochondria in the liver of ALS patients32,56. These findings support the evidence that mutant ER-stress regulating genes such as XBP1, SigR1, VCP, TDP-43, FUS, SOD1, and VAPB are linked to ALS57,58. Furthermore, SMN gene, implicated in ALS and SMA, have been shown to regulate the development and function of liver35.
Finally, hepatic steatosisis is linked to the metabolic syndrome, characterised by hyperglycaemia, hyperglucagonemia, insulin resistance and altered serum triglycerides; and such findings have been reported in ALS59–65. In this regard, it is interesting to note that damage to fast twitch skeletal muscle, the main site of glucose disposal and the largest reservoir of glycogen in humans, leads to hepatic steatosis66.
Notably, Li et al. showed exendin-4, which counteracts hepatic steatosis, ameliorated motor neuron degeneration partly by correcting this systemic metabolic alteration67,68. This clearly suggests that, much like skeletal muscle, liver pathology is not merely an innocent bystander, but rather a premorbid condition, which plays an active role in ALS pathogenesis.
Aims
Thus, (i) identifying skeletal–muscle produced unknown pathological factor, (ii) unravelling its nexus and synergism with hepatic steatosis, (iii) understanding the mechanisms by which this pathology factor causes motor neuron damage, and (iv) revealing the cause(s) of clinical heterogeneities would fully untie the Gordian knot of ALS pathology, allowing the development of predictive and prognostic biomarkers as well as potent drugs3,13. Hence, by taking a systems view, this paper aims to fill these knowledge gaps. Moreover, by fusing these separate pieces together, this paper presents a full picture of ALS pathology.
Impaired glycolysis in fast twitch muscle: one of the pathological triggers of ALS
Evidence suggests that defective energy deficit in skeletal muscle triggers ALS. Investigators found impaired skeletal muscle metabolism, characterised by low ATP levels and hypermetabolism, causes neuromuscular dysfunction in ALS mouse model69,70. Conversely, metabolic interventions such as high-calorie diets and reducing hypermetabolism improved survival and alleviated symptoms in ALS19,70. However, the functional link between skeletal muscle metabolic impairment and ALS remains nebulous. Instructively, since ALS begins from fast twitch muscle, which relies on anaerobic glycolysis for energy (i.e., ATP), this immediately suggests that impaired anaerobic glycolysis produces the unknown pathological trigger.
Impaired glycolysis in ALS
Compelling evidence suggests that muscle glycolysis is impaired in ALS. Valosin-containing protein (VCP), a gene linked to ALS, causes defective muscle glycolysis and reduced ATP levels. Dupis et al. linked upregulation of mitochondrial uncoupling proteins UCP1 and UCP3—which suppresses glycolysis and causes hypermetabolism—to muscle denervation in ALS71. Bernardini et al. showed low expression of glycolysis genes such as FBP2 and enolase 3 in the skeletal muscles of ALS patients72. Brockington et al. uncovered down regulation of glycolytic enzyme lactate dehydrogenase 1 in the VEGFδ/δ mouse model of ALS73. Moreover, experiments showed that the gain-of-interaction of the SOD1G93A mutant with cytosolic malate dehydrogenase induces glycolytic impairments74. Dunckley et al. linked variants of FLJ10986, a protein linked to glycolysis, with the susceptibility of sporadic ALS75. Collectively, these findings clearly show impaired glycolysis in skeletal muscle of ALS patients and mouse model.
Impaired muscle glycogen and glucose homeostasis in ALS
Notably, fast twitch skeletal muscle glycolysis depends on muscle glycogen storage and glucose transporter 4 (GLUT4)-mediated muscle glucose uptake26,76. Accumulating evidence suggests defective muscular glycogen metabolism and impaired GLUT4-mediated muscular glucose uptake in ALS. Derave et al. discovered diminished muscle ATP and glycogen accumulations in SOD1 G93A mice27. Smittkamp et al. revealed impaired insulin-stimulated glucose uptake exclusively in fast twitch skeletal muscle in middle-stage SOD1 G93A mice77. Accordingly, fast twitch skeletal muscle fibres of TDP-43 transgenic mice show defective insulin-induced GLUT4 translocation and glucose uptake77. Moreover, in the mutant TDP-43-linked ALS mice, Perera et al. reported decreased AMPK, which mediates muscle contraction-induced glucose entry and glycogen synthesis76,78,79. Conversely, AMPK activator drugs (i.e. latrepirdin) delayed ALS in SOD1G93A mice80. Furthermore, muscle contraction facilitated glucose uptake involving Ca2+/calmodulin-dependent GLUT4 translocation appears to be defective in ALS. For example, investigators linked mutant neuregulin-ERBB4 gene, involved in calcium-induced glucose uptake during muscle contraction, to ALS79,81. Thus, it is obvious that ALS involves impaired carbohydrate metabolism that supports muscle glycolysis.
ALS resistance of extraocular muscles (EOMs): role of glycolysis
Finally, the metabolic characteristics of—ALS-resistant—extraocular muscles (EOMs) further consolidate this notion82. Two fundamental differences exist between EOMs and skeletal muscle metabolism83. First, compared to skeletal muscles, EOMs have high glycolysis capacity, evident by the overexpression of glycolytic enzymes (e.g. lactate dehydrogenase, enolase)83. Second, owing to their high vascularity, EOMs rely more on instantaneous glucose uptake—less on glycogen storage and GLUT4-mediated muscle glucose uptake83. All in all, these three sets of findings point that defective glycolysis causes ATP deficits in fast twitch skeletal muscle of ALS patients. Hence, the unknown pathological factor emanating from skeletal muscle appears to have direct connection with defective muscle glycolysis. How?
Ammonia: the elusive pathological factor
Notably, defective glycolysis, which reduces ATP levels, in fast twitch skeletal muscle activates catabolic reactions of adenine nucleotides (i.e. purine nucleotide cycle) and amino acids (Figure 1)84,85. Intriguingly, such catabolic reactions produce ammonia—a neurotoxin 1000 times more toxic than ethanol at equimolar concentrations85,86. Since ammonia is toxic, it is obligatorily removed mainly through hepatic urea cycle which transforms ammonia into urea87. Notably, when the urea cycle is impaired, as it occurs in fatty liver disease, increased ammonia production from skeletal muscle or from dietary sources can cause chronic hyperammonia (>35–50 µM) and consequent neurodegeneration and motor impairments (Figure 1 and Figure 2)33,88–92. Indeed, in many liver diseases, including fatty liver disease which commonly occurs in ALS, because of impaired urea cycle-mediated ammonia removal, hyperammonia frequently leads to corticospinal hyperexcitability, myelopathy and spasticity—features strikingly reminiscent of neurophysiological phenotypes of ALS symptoms93–97.
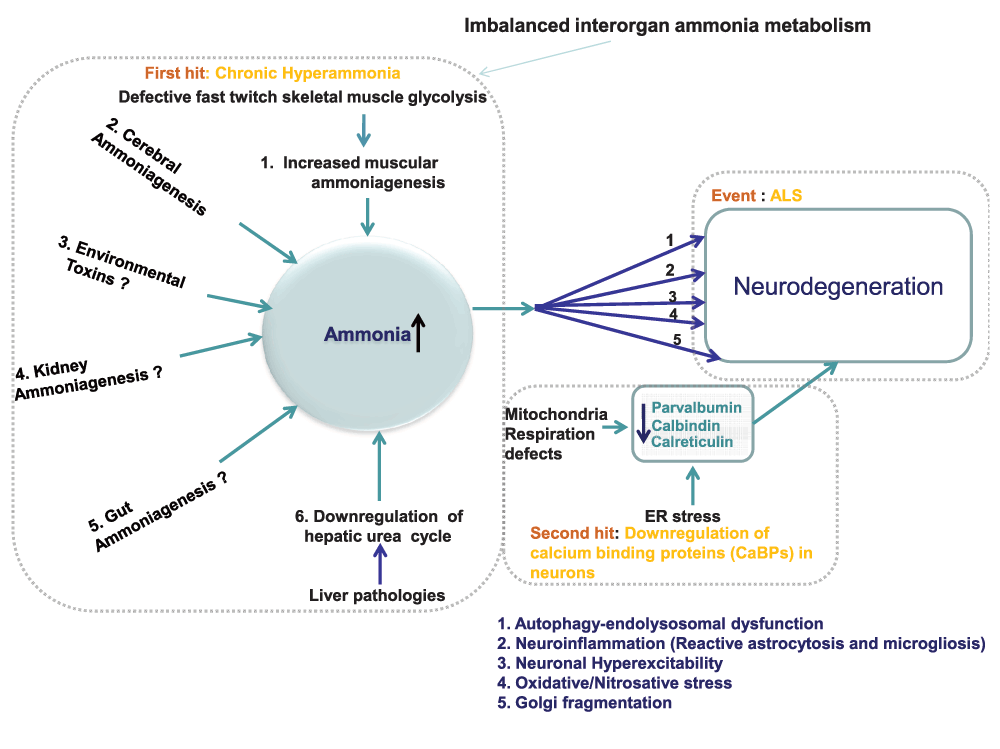
Figure 1. Line diagram: mechanisms of motor neuron damage in ALS.
Mechanism of motor neuron degeneration in ALS involves two main factors: (i) ammonia neurotoxicity and (ii) down regulation of neuronal calcium binding proteins (CaBPs). Owing to imbalanced interorgan ammonia metabolism, ammonia, a well-known neurotoxin, accumulates in neurons. Among the five organs (brain, skeletal muscle, gut, liver and kidney) involved in ammonia metabolism, ALS appears to mainly involve the role of liver and skeletal muscle in that confluence of impaired ammonia removal—owing to impaired hepatic urea cycle—and increased muscular ammoniagenesis—owing to impaired glycolysis in fast twitch skeletal muscle—lead to chronic hyperammonia in ALS. In the brain, ammonia activates several neurodegenerative pathways such as (1) autophagy-endolysosomal dysfunction (2) neuroinflammation (3) oxidative stress (4) Golgi fragmentation and (5) neuronal hyperexcitability. In the absence of neuronal calcium binding proteins (CaBPs) such as parvalbumin, calbindin, calreticulin, activation of these degenerative pathways lead to motor neuron damage. Notably, decrease in calreticulin, because of increased ER stress, leads to lower motor neuron damage, whereas the down-regulation of parvalbumin and calbindin, because of defective mitochondrial respiration, leads to upper motor neuron damage.
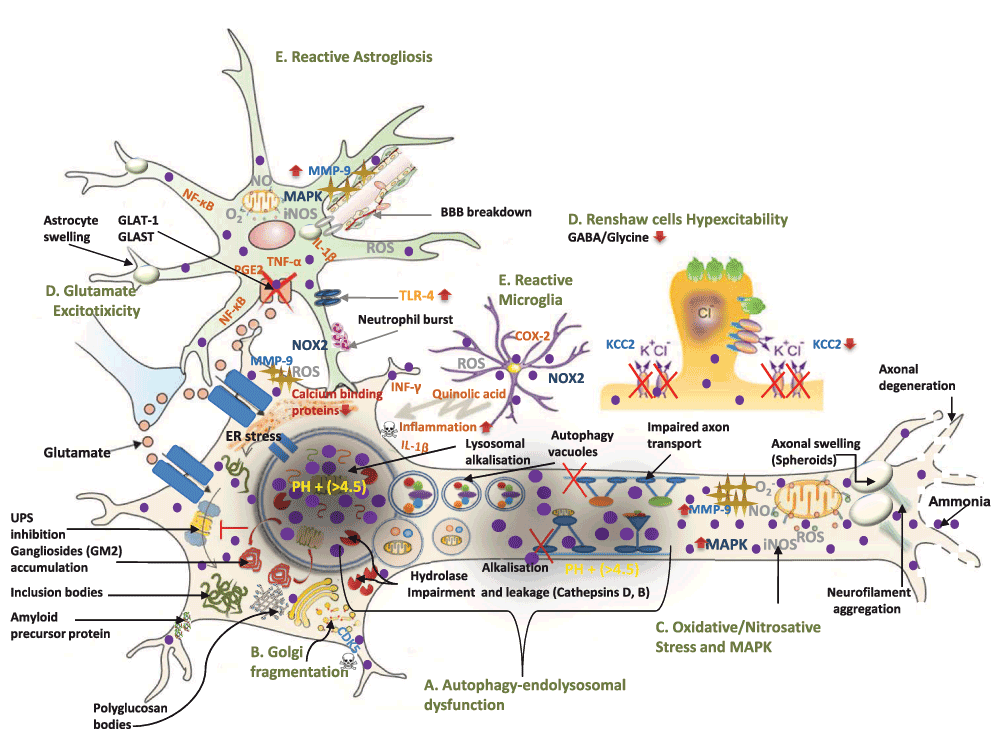
Figure 2. A molecular model of ammonia-induced motor neuron degeneration in ALS.
(Modified with permission from 232). Ammonia intoxication directly damages motor neurons through five mutifactorial pathological mechanisms: 1) alkalisation-induced impairment of macroautophagy-endolysosomal system, 2) Golgi impairment, 3) increased oxidative/nitrosative stress and MAPK up-regulation 4) neuronal hyperexcitability and 5) neuroinflammation. These mechanisms explain frequently found cellular, molecular and neurophysiological phenotypes of motor neuron damage in ALS. A. Owing to ammonia-induced alkalisation, impairment of macroautophagy-endolysosomal system induces several key molecular histopathological features of ALS including : (i) ubiquitinated (Lewy and skein body-like inclusions) and non-ubiquitinated inclusion bodies (i.e. bunia bodies) formation, (ii) amyloid precursor protein (APP), (iii) gangliosides accumulation (i.e. GM2), (iv) autophagy vacuoles, (v) neurofilament aggregation and axonal swelling. B. Ammonia activates CDK5 which in turn leads to frequently observed Golgi fragmentation. C. Ammonia-induced oxidative/nitrosative stress and MAPK-up-regulation lead to multiple cellular and molecular pathological features such as: (i) blood brain barrier (BBB) breakdown, and (ii) MMP-9-induced ER stress. D. Ammonia causes neuronal hyperexcitability by (i) down-regulating astrocyte glutamate transporters (GLAT-1 and GLAST) and (ii) lowering potassium-chloride co-transporter KCC2 level which suppresses GABA and Glycine-mediated inhibitory neurotransmission. E. Ammonia leads to neuroinflammation secondary to reactive microglial and astrogliosis. This occurs because of (i) quinolic acid release from microglia (ii) up-regulation of pro-inflammatory cytokines (TNF, IL-1β, NF-κB, and PGE2) in astrocytes (iii) TLR-4 activation and (iv) neutrophil burst derived NADPH oxidase (NOX)-induced oxidative stress.
Ammonia neurotoxicity: hypothesis and evidence
Together, these findings provide a compelling rationale for a new hypothesis. ALS pathology might involve not only skeletal muscle-induced increased ammonia production, because of impaired glycolysis, but also impair ammonia removal, secondary to hepatic steatosis-induced faulty urea cycle, leading to chronic hyperammonia and consequent progressive motor neuron degeneration (Figure 1 and Figure 2). Astonishingly ammonia’s role has seldom been directly investigated. Nonetheless, diverse data obtained from clinical and animal studies support this hypothesis showing that hyperammonia increases ammoniagenesis and decreases ammonia removal in ALS.
A clinical study showed elevated ammonia level in motor neuron disease patients—with ammonia levels inversely correlated to disease duration98. These investigators also found a causal relationship between ammonia and ALS by noting that infusion of amino acids, which causes ammoniagenesis, aggravates ALS98. Moreover, dietary supplements of branched chain amino acids was one of the factors associated with the early onset of ALS (45 years) in Italian soccer players99. Accordingly, other investigators reported accelerated skeletal muscle protein catabolism ALS100. This chimes with the fact that hepatic steatosis-induced glucagon secretion, which occurs in ALS, increases ammoniagenesis through protein degradation101,102. Consistent with this, as noted above, intense or prolonged physical exertion, an ammoniogenic activity, is an ALS risk factor11,103.
Beside a link between hepatic steatosis and a faulty urea cycle, other lines of clinical evidence further implicate the impaired urea cycle in ALS48,91,104. For example, Iłzecka et al. showed decreased arginine levels, an amino acid required for liver urea cycle function, in ALS patients105. Additionally, research showed that metabolic acidosis, which impairs urea cycle, occurs in ALS60,106. Impaired hepatic urea cycle activates glutamine synthetase, an alternative ammonia detoxification pathway, and researcher also found increase in glutamine synthetase expression in blood platelets of ALS patients107,108.
Animal models of ALS further cement this ammonia hypothesis. Investigators showed hyperammonia and impaired urea cycle in 50 day old SOD1G93A mice compared to wild type mice of the same age109. Moreover, these investigators showed increased glutamine, a precursor of ammonia, in SOD1G93A mice109. Additionally, in the mutant SOD 1 G86R mice, de Aguilar et al. showed early (3 months of age) muscle denervation along with increased AMP deaminase-3 (AMPD3), an enzyme of purine nucleotide cycle involved in ammoniagenesis110. Furthermore, a set of studies showed increased arginine vasopressin release in the SOD1 mice, and independent research showed that arginine vasopressin causes muscle protein degradation and consequent ammoniagenesis111,112. Conversely, research showed that ammonia-counteracting compounds such as phenylbutyrate, ariginine, resveratrol and l-carnitine alleviated symptoms and enhanced survival in the ALS mouse model97,113–118.
Further supporting this hypothesis, experiments have shown that environmental neurotoxins implicated in ALS causes ammonia toxicity. Dietary intake of β-N-methylamino-L-alanine, a non-protein amino acid linked to Guam's ALS-PDS complex epidemic, causes liver damage and ammonia toxicity119,120. Similarly, the ingestion of Lathyrus sativus seeds, implicated in neurolathyrism (an upper motor neuron disease), causes liver dysfunction, urea cycle impairment, and chronic ammonia toxicity121,122. Finally, animal studies showed that the pesticide pyrethroid, which causes an ALS-mimicking syndrome, leads to protein catabolism ammonia toxicity123,124.
Yet another line of evidence bolsters the ammonia neurotoxicity hypothesis. Interestingly, reports showed that motor neuron disease could be one of Huntington's disease (HD)’s presenting features125,126. In fact, aside from genetic overlap with ALS, HD shares many pathophysiological characteristics with ALS: skeletal muscle atrophy, hepatic steatosis, hyperglycaemia and adipose tissue dysfunction127,128. Tellingly, although often regarded as curious findings rather than telltale observation, impaired urea cycle as well as hyperammonia occur in HD127. Strikingly, data from mouse models of HD showed that protein-restricted diets not only reduced hyperammonia but also prevented the motor deterioration90. This suggests that ammonia could be a common culprit in range of neurodegenerative conditions, especially affecting motor system.
In addition to muscles and the liver, ammonia metabolism involves other organs, including the gut, the kidneys and the brain (Figure 1)87,89. Hence, this hypothesis does not preclude a role of these organs. Although no evidence has yet emerged to implicate the gut and kidneys in ALS, some data at least suggest a role of cerebral ammoniagenesis in ALS. Studies showed increased deamination of catecholamine, which causes cerebral ammoniagenesis, in ALS, evident by the overactivity of catecholamine oxidising enzymes such as MAO-B and aldehyde oxidase89,129–131. Put together, these findings implicate ammonia neurotoxicty in ALS.
Mechanisms of ammonia’s neurotoxicity
When hyperammonia occurs, ammonia enters into the brain, leading to neurotoxicity. Ammonia exerts pleiotropic neurotoxic effects by activating an array of cellular mechanisms, which are the proximal causes of ALS (Figure 1 and Figure 2). These mechanisms include: 1) alkalisation-induced impairment of macroautophagy-endolysosomal system, 2) Golgi impairment, 3) increased oxidative/nitrosative stress and mitogen-activated protein kinase (MAPK) up-regulation 4) neuronal hyperexcitability and 5) neuroinflammation89,132–136.
As described below, taken together, these five mechanisms not only explain several frequent cellular and molecular histopathological hallmarks of ALS but also neurophysiological features of ALS (Figure 1 and Figure 2). The cellular histological features, explained by ammonia’s toxicity, include axon swelling, blood brain barrier breakdown and astrogliosis and microgliosis137–139. The molecular pathological features, explained by ammonia’s toxicity, include formation of inclusion bodies such as bunia bodies and Lewy bodies, gangliosides accumulation, glycogen aggregation, neurofilament derangement, Golgi fragmentation, and reduced glutamate transporters60,140–145. Moreover, ammonia toxicity explains a key neurophysiological feature of ALS: neuronal hyperexcitability17. Finally and most importantly, ammonia toxicity explains why ALS is mainly a motor neuron disease.
Alkalisation-induced impaired macroautophagy-endolysosomal system
Ammonia-induced alkalisation impairs the macroautophagy-endolysosomal system, one of the main cellular garbage disposal systems. This occurs at least in two ways. First, ammonia, a weak base, preferentially accumulates in lysosomes because of their low acidity (PH~4.5)134. Consequently, intra-lysosomal alkalisation and lysosomal enzyme leakages occur, impairing the lysosomal hydrolysis of proteins, lipids and carbohydrates (Figure 2)134. Second, ammonia alkalises acidic membranous compartments of axon terminals, jamming membrane microtubules and thereby blocking the anterograde-to-retrograde transport of endosomes146. Consequently, impaired fusion of endocytic compartments with lysosomes occurs, causing defective autophagy of endocytosed material (Figure 2)89,146,147. As a result, toxic accumulation of protein aggregates, glycolipids, and carbohydrates occurs89. In turn, these toxic by-products activate the apoptosis programme, causing cell death148.
Ammonia’s alkalisation-induced toxicity is especially relevant to ALS because macroautophagy-endolysosomal dysfunction causes motor neuron degeneration149. Indeed, mutant genes of this pathway such as SOD1, FIG4, CHMP2B, SQSTM1, DCTN1, DYNC1H1, and RAB7A have been linked to ALS149. Consistent with this interpretation, research showed impaired dynein-dependent retrograde axonal transport, required for autophagosome-lysosome fusion, causes motor neuron degeneration150,151. Furthermore, consistent with lysosomal enzyme leakage, investigators reported increased lysosomal enzyme levels (i.e. acid phosphatase, Cystatin C) in the cerebrospinal fluid (CSF) and plasma of ALS patients152,153.
Impaired lysosomal proteolysis
Ammonia-induced impaired lysosomal proteolysis explains key histopathological hallmarks of ALS including formation of inclusion bodies (Figure 2). Ammonia-induced alkalinisation in lysosomes impairs the activities of protease enzymes including cathepsin B and cathepsin D134,154. Strikingly, investigators showed downregulation of cathepsin B and cathepsin D in ALS155,156. Notably, defective lysosomal proteolysis causes swollen axonal dystrophy (spheroids), with histological features such as ubiquitinated and non-ubiquitinated inclusion bodies, amyloid precursor protein, and neurofilament aggregation157. In keeping with this, research revealed such findings in ALS144,145,156,158–161.
In regard to non-ubiquitinated inclusion bodies, Kikuchi et al. showed that decreased cathepsin B generates Bunina bodies (small eosinophilic intraneuronal lysosomal inclusion bodies) in motor neurons, a hallmark of ALS (Figure 2)140,156. Since cathepsin D mediates lipofuscin and α-synuclein clearance, and since downregulation of cathepsin D occurs in ALS, this explains frequently observed deposits of lipofuscin granules and α-synuclein aggregation in ALS patients148,162–164. Moreover, reduced cathepsin B activity induces amyloid precursor protein (APP) accumulation, and Bryson et al. showed increased APP level in the SOD1 G93A mouse, which contributed to motor neuron damage158,165. As for impaired proteolysis-induced ubiquitinated inclusion bodies, ubiquitin inclusion aggregates such as Lewy body-like inclusions’ and ‘skein-like inclusions’ have been found in ALS (Figure 2)145,166. This finding accords with the observations that inhibition of macroautophagy impairs the ubiquitin proteasome system (UPS)167. Finally, neurofilament aggregation and spheroid formations have been found in the ALS mouse model and in patients (Figure 2)161.
Impaired lysosomal ganglioside clearance
Gangliosides are complex sialylated glycosphingolipids, particularly found in the CNS168. Notably, GM2 ganglioside is a main ganglioside in motor neurons169. Accumulation of GM2 ganglioside, owing to impaired lysosomal Hexosaminidase (Hex) enzymes, frequently causes motor neuron disease170–172. For example, Banerjee et al. reported slow accumulation of GM2 ganglioside, primarily in motor neurons, in patients with progressive motor neuron disease associated with partial Hex A and no Hex B activity172. By implication, this suggests that accumulation of gangliosides including that of GM2 occurs in ALS and that ammonia increases GM2 ganglioside levels. Indeed, although scantly investigated, some investigators reported increased ganglioside levels in ALS including GM2 ganglioside142,173,174. In line with ammonia’s role in ganglioside metabolism, Perez et al. showed that ammonia causes leakage of Hexosaminidase A (Hex A), indicating GM2 accumulation175,176. Thus, ammonia-induced GM2 accumulation could partly explains the heightened vulnerability of motor neurons in ALS (Figure 2).
Impaired lysosomal carbohydrate clearance
Animal and clinical studies reported neuronal and glial glycogen accumulation and polyglucosan bodies (branched chained glycogen aggregates) in ALS (Figure 2)60,177,178. Notably, Dodge et al. showed that decreased level of α-glucosidase—a glycogen degrading lysosomal enzyme—partly causes glial and neuronal glycogen accumulation in ALS, and experiments showed that ammonia leaks α-glucosidase from lysosomes60,179. Thus, ammonia-mediated lysosomal dysfunction explains yet another histological feature of ALS. Of note, this fits with the observations that upper and lower motor neuron lesions frequently arise in polyglucosan body diseases180.
Impaired Golgi function
Ammonia toxicity could explain Golgi apparatus fragmentation in ALS, an early and frequently observed event141. Sun et al. showed that CDK5 activation fragments Golgi apparatus181. Interestingly, Cagnon and Braissant showed that ammonia activates CDK5. They also showed that CDK5 activation led to neuronal cell death and impairment of axonal outgrowth135. Apparently, p25-induced mislocalization and deregulation of CDK5 activity occurs in ALS (Figure 2)143,182. In fact, Nguyen et al. reported that an attempted re-entry of motor neurons into the G1-S phase of the cell cycle subsequent to CDK5 deregulation is a critical step of neurodegeneration in ALS182.
Increased oxidative/nitrosative stress and MAPK expression
Additionally, data suggested that ammonia induces oxidative/nitrosative stress and MAPK expression, frequently found pathological features of ALS (Figure 2)132. Research showed that oxidative/nitrosative stress and MAPK increases extracellular matrix degrading enzymes such as urokinase-type plasminogen activators and MMP-9183. Unsurprisingly, experiments found that increased levels of these extracellular matrix degrading enzymes occur in ALS184. Strikingly, Kaplan et al. observed overexpression of MMP-9 increased the vulnerability of fast fatigable limb-innervating motor neuron185. MMP-9 appears to exert neurotoxicity mainly through up-regulation of ER stress (Figure 2)185. Moreover, Skowrońska et al. showed that increase in MMP-9, which degrades the extracellular matrix, destroys the blood brain barrier (BBB)186. Predictably, Nicaise et al. showed impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rodents138. Additionally, since MAPK regulates cytoskeletal homeostasis, ammonia-induced MAPK activation explains why cytoskeleton abnormalities such as intermediate filaments accumulation occur in ALS160,187.
Neuronal hyperexcitability
Furthermore ammonia intoxication explains neuronal hyperexcitability in ALS—a cardinal characteristic of ALS16. By decreasing potassium-chloride cotransporter KCC2, located in the brain and spinal cord, ammonia increases chloride levels in neurons (Figure 2)188. Increased neuronal chloride levels in turn suppress GABA and Glycine-mediated inhibitory neurotransmission, causing neuronal hyperexcitability (Figure 2)189. In keeping with this, Fuchs et al. discovered decreased KCC2 expression in ALS-vulnerable motoneurons in spinal cord and hypoglossal nuclei of SOD1-G93A mice but not in EOMs190. Concordantly, researchers reported spinal motor neuron hyperexcitability and degeneration in ALS patients191. In fact, Hübner et al. showed that KCC2 knockout mice died after birth owing to motor deficits that caused respiratory failure, a feature similar to ALS189.
Furthermore, ammonia causes glutamatergic excitotoxicty. By MAPK activation and increasing oxidative stress, ammonia decreases the glutamate transporter EAAT2 (GLT-1) and glutamate-aspartate transporter (GLAST) (EAAT-1) in astrocytes (Figure 2)133,192. Consequently, decreased transporters impair astrocyte-mediated high affinity glutamate uptake and clearance, leading to defective glutamatergic neurotransmission and excitotoxicity133,192,193. In line with this, decreased GLT-1 and GLAST have been found in the spinal cord of SOD1 G93A mice and ALS patients194–196. Interestingly, increased CSF glutamate was associated with a spinal onset of the disease and with severity of the symptoms in 41% of ALS patients196.
Neuroinflammation
Ammonia extensively affects the function of astrocytes and microglia (Figure 2). Through several mechanisms including (1) quinolinic acid (QUIN) production, (2) NADPH oxidase (NOX) activity-induced reactive oxygen species (ROS) generation, (3) Toll-like receptor 4 (TLR-4) activation, and (4) extracellular-signal-regulated kinase (ERK) pathway stimulation, ammonia induces a transition from a resting state into reactive astroglia and microglia phenotype (Figure 2)113,197–199. Consequently, reactive astroglia and microglia increase oxidative stress and stimulate the release of a range of proinflammatory cytokines including NF-κB, IL-1β, and PGE2, leading to neuroinflammation and degeneration (Figure 2)200.
Emerging data indicate a role of QUIN in ALS201. Chen et al. detected overproduction of serum tryptophan, kynurenine and QUIN in the CSF of ALS patients compared to controls, concomitant with microglial activation and neuroinflammation (Figure 2)139. Similarly, experiments showed increased microglial and neutrophil-derived NOX activity correlated with fast ALS progression202. In keeping with increased ammonia-induced inflammation, investigators showed TLR-4 activation, and elevated levels of various pro-inflammatory cytokines in ALS203,204.
Clinical heterogeneities in ALS: the role of calcium-binding proteins (CaBPs)
The postulated ammonia neurotoxicity as the sole cause of ALS raises an awkward question. If ammonia damages both upper and lower motor neurons equally, then why does ALS often deviate from its classical pattern, manifesting as either the upper or lower motor neuron dominant subtype8? Moreover, why it is a relatively rare disorder? These questions clearly indicate that a protective factor exists that counteracts ammonia toxicity, and that anatomic-region specific loss of this factor causes the clinical heterogeneities in its presentation.
One such neuroprotective factor identified in ALS is the ER family of calcium binding proteins (CaBPs) (Figure 1 and Figure 2)205. By regulating voltage-gated calcium ion channels, CaBPs reduce calcium overload and cytotoxicity, thus protecting neurons from cell death205. The CaBPs involved in motor neuron protection include calreticulin, parvalbumin, and calbindin which are distributed in anatomic region specific manner within motor neurons206,207. Calreticulin expression mainly occurs in limb-innervating lower motor neuron regions such as the lumbar spinal cord area and fast-fatigable motoneuron, whereas calbindin and parvalbumin are expressed in both lower and upper motor neurons206,207.
Differential anatomic region-specific distribution of CaBPs in the CNS partly explains different patterns of motor neurodegeneration206,208. In the SOD1 G93A ALS mouse model, during the presymptomatic stage, fast-fatigable motoneuron denervation mainly accompanies calreticulin loss208,209. By contrast, investigators showed that loss of calbindin and parvalbumin correlated with both upper and lower motor neuron damage206.
Region specific regulation of CaBPs: ER stress and bioenergetics
How do neurons lose different CaBPs in different anatomic regions of the CNS? Research showed that ER stress downregulates calreticulin in limb-innervating lower motor neurons motor. In fact, calreticulin co-localises with the ER207. This accords with the finding that neuronal MMP-9—which enhances ER stress—selective damages fast fatigable lower motor neurons185. Additionally, since androgens modulate ER stress, this explains why sexual dimorphism occurs in lower motor neuron damage210. Interestingly, research revealed increased ER stress and reduced calreticulin in Alzheimer’s disease (AD)209. This explains why AD occasionally co-exists with motor neuron disease211.
As for the causes of reduced parvalbumin and calbindin expression in ALS, research implicates impaired oxidative metabolism secondary to defective mitochondrial electron transport (the respiratory chain) system212,213. Indeed, of the five protein complexes of the mitochondrial respiratory chain, research has frequently showed reduced respiratory chain complex I and IV activity in sporadic ALS patients205,212. Within these two complexes, complex IV appears to be particularly involved in ALS. This chimes well with the fact that 90% of all parvalbumin and calbindin-immunoreactive cells showed dense staining for respiratory complex IV (cytochrome c oxidase)214. Furthermore, hyperhomocysteinaemia, found to be highly prevalent in ALS, damages mitochondria and suppresses respiratory complex IV activity37,215. Revealingly, compared to skeletal muscle, the EOMs have slow metabolism characterised by low complexes I and IV activities (~50%) yet elevated mitochondria density with increased complex I and IV levels (30% to 2 times)—explaining why parvalbumin and calbindin levels remain relatively unaffected in EOMs205,216,217.
The role of respiratory chain complex subunits
Interestingly, alterations in mitochondrial respiratory chain complex subunits also partly determine the spectrum of motor neuron damage. Investigators reported that altered Cytochrome c oxidase subunit Vb caused spinobulbar muscular atrophy, whereas Cytochrome c oxidase subunit I microdeletion induced upper motor dominant motor neuron damage218,219. Furthermore, deficiency of complex I involved lower motor neuron damage involving spinal and bulbar areas220. This fits with the findings that anatomic region-specific differences in mitochondrial respiration contribute to the localized neurodegeneration221.
In summary, these findings suggest that the regional loss of CaBPs expression, dependent on ER stress and defective mitochondrial respiration in the brain determines the anatomically variable manifestation of ALS. Collectively, it is also clear that motor neuron degeneration depends not only on postulated ammonia neurotoxicity but also on deficits of CaBPs within motor neuron.
Biomarkers and therapeutics
From this insight about ALS pathogenesis, diagnostic, disease monitoring and therapeutic measures emerge—fostering real hopes that ALS can be halted or even cured. Because ammonia is a volatile organic compound, excreted from breath and skin, an ammonia breath test would present a simple, reliable, robust, inexpensive and non-invasive tool for diagnosis and monitoring of ALS222. This ammonia breath test would prove invaluable in expediting drug discovery process. Aside from ammonia, gangliosides (e.g. Sialosylglobotetraosylceramide) and serum lysosomal enzymes could also serve as reliable adjuvant biomarkers of ALS142.
As for therapeutics, since ammonia toxicity appears to be a major player in ALS, ammonia-removal strategies seem to be the most effective strategy for ALS treatment223. Many existing ammonia-lowering agents including those that act on the hepatic urea cycle can be employed224,225. These could include salbutamol, conclevan, neomycin, sodium benzoate, ornithinephenyl acetate and L-ornithine aspartate223,226,227. Moreover, since impaired fast-twitch skeletal muscle glycolysis plays a role in ALS, improving muscle glycolysis through various existing drugs such as serotonin agonists and AMPK agonists (e.g. D-xylose) is another promising pharmacological strategy228,229. Additional therapeutic strategies could involve correcting system metabolic defects such as hyperglucagonemia and acidosis60,68. Moreover, other potent therapeutic targets could involve MAPK inhibitors, K-Cl co-transporters, and hexosaminidase agonists (e.g. Pyrimethamine)192,230,231. Finally, interventions that restore the levels of CaBPs should also be simultaneously applied for effective treatment.
Summary
ALS is a ghastly and incurable disease. Despite increasing wealth of data, ALS remains poorly understood. By analysing existing literature, this paper has not only identified important knowledge gaps in ALS aetiopathology but also filled them and tied them together. In doing so, this paper postulates a new integrative explanation of ALS and suggests potent therapeutic measures to treat ALS. Central to this explanation is the notion that ALS is a neurological disease of metabolic origin—resembling hepatocerebral degeneration223. This explanation posits that ALS pathology involves the interplay of two critical factors: 1) chronic hyperammonia caused by imbalanced interogan ammonia metabolism, mainly due to muscle and liver pathology (Figure 1 and Figure 2) altered CaBPs homeostasis, mainly due to increased ER stress and impaired mitochondrial respiration (Figure 1).
Considering all these together in sequence, impaired fast twitch skeletal muscle carbohydrate metabolism activates purinergic and amino acid catabolism, leading to a release of ammonia, a neurotoxin. Alternatively, ammonia toxicity can also be induced or exacerbated by other endogenous (e.g. cerebral deamination, intestinal ammoniagenesis) and exogenous sources (i.e. neurotoxins). Owing to concurrent liver pathology (e.g. hepatic steatosis) in ALS, impaired hepatic ammonia detoxification occurs. Consequently, ammonia levels progressively builds up, leading to chronic hyperammonia. Since ALS pathology also involves loss of neuroprotective CaBPs (i.e. calbindin, calreticulin and parvalbumin), ammonia neurotoxicity in the absence of CaBPs leads to ALS. Ammonia damages motor neurons through a range of pathways. These pathways include impaired macroautophagy-endolysosomal impairment, Golgi fragmentation, oxidative/nitrosative stress and reactive microglial and astrogliosis. These mechanisms explain a range of histopathological and neurophysiological hallmarks of ALS such as bunia bodies and neuronal hyperexcitability. Finally, since ALS appears to be associated with HD, dementia and Parkinsonism this framework can be generalised to explain these disorders33,89,90.
Competing interests
No competing interests were disclosed.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Acknowledgements
I am very grateful to Prof. Rod Nicolson, at the University of Sheffield, for his kindness, constant support, and editorial guidance during this work. I am thankful to Prof. Paul Overton, at the University of Sheffield, for his incisive comments and criticisms. I am also thankful to Navin Shah and Nirav Parekh for their support.
Faculty Opinions recommendedReferences
- 1.
Maessen M, Veldink JH, Onwuteaka-Philipsen BD, et al.:
Euthanasia and physician-assisted suicide in amyotrophic lateral sclerosis: a prospective study.
J Neurol.
2014; 261(10): 1894–901. PubMed Abstract
| Publisher Full Text
- 2.
Carus R:
Motor neurone disease: a demeaning illness.
Br Med J.
1980; 280(6212): 455–456. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 3.
Henriques A, Gonzalez De Aguilar JL:
Can transcriptomics cut the gordian knot of amyotrophic lateral sclerosis?
Curr Genomics.
2011; 12(7): 506–515. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Stone N:
Amyotrophic lateral sclerosis: a challenge for constant adaptation.
J Neurosci Nurs.
1987; 19(3): 166–73. PubMed Abstract
- 5.
Bastos AF, Orsini M, Machado D, et al.:
Amyotrophic lateral sclerosis: one or multiple causes?
Neurol Int.
2011; 3(1): e4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Turner MR, Swash M, Ebers GC:
Lockhart Clarke’s contribution to the description of amyotrophic lateral sclerosis.
Brain.
2010; 133(11): 3470–3479. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Pansarasa O, Rossi D, Berardinelli A, et al.:
Amyotrophic lateral sclerosis and skeletal muscle: an update.
Mol Neurobiol.
2014; 49(2): 984–90. PubMed Abstract
| Publisher Full Text
- 8.
Swinnen B, Robberecht W:
The phenotypic variability of amyotrophic lateral sclerosis.
Nat Rev Neurol.
2014; 10(11): 661–670. PubMed Abstract
| Publisher Full Text
- 9.
Ferraiuolo L, Kirby J, Grierson AJ, et al.:
Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis.
Nat Rev Neurol.
2011; 7(11): 616–30. PubMed Abstract
| Publisher Full Text
- 10.
Chen S, Sayana P, Zhang X, et al.:
Genetics of amyotrophic lateral sclerosis: an update.
Mol Neurodegener.
2013; 8(1): 28. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Ferraiuolo L, De Bono JP, Heath PR, et al.:
Transcriptional response of the neuromuscular system to exercise training and potential implications for ALS.
J Neurochem.
2009; 109(6): 1714–24. PubMed Abstract
| Publisher Full Text
- 12.
von Giesen HJ, Kaiser R, Köller H, et al.:
Reversible ALS-like disorder in HIV infection. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy.
Neurology.
2002; 59(3): 474; author reply 474–5. PubMed Abstract
- 13.
Turner MR, Bowser R, Bruijn L, et al.:
Mechanisms, models and biomarkers in amyotrophic lateral sclerosis.
Amyotroph Lateral Scler Frontotemporal Degener.
2013; 14(Suppl 1): 19–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
Dadon-Nachum M, Melamed E, Offen D:
The "dying-back" phenomenon of motor neurons in ALS.
J Mol Neurosci.
2011; 43(3): 470–7. PubMed Abstract
| Publisher Full Text
- 15.
Sargsyan SA, Monk PN, Shaw PJ:
Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis.
Glia.
2005; 51(4): 241–53. PubMed Abstract
| Publisher Full Text
- 16.
Shaw PJ, Ince PG:
Glutamate, excitotoxicity and amyotrophic lateral sclerosis.
J Neurol.
1997; 244(Suppl 2): S3–14. PubMed Abstract
| Publisher Full Text
- 17.
Ince PG, Lowe J, Shaw PJ:
Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology.
Neuropathol Appl Neurobiol.
1998; 24(2): 104–17. PubMed Abstract
| Publisher Full Text
- 18.
Bruijn LI, Miller TM, Cleveland DW:
Unraveling the mechanisms involved in motor neuron degeneration in ALS.
Annu Rev Neurosci.
2004; 27: 723–49. PubMed Abstract
| Publisher Full Text
- 19.
Dupuis L, Pradat PF, Ludolph AC, et al.:
Energy metabolism in amyotrophic lateral sclerosis.
Lancet Neurol.
2011; 10(1): 75–82. PubMed Abstract
| Publisher Full Text
- 20.
Ilieva H, Polymenidou M, Cleveland DW:
Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond.
J Cell Biol.
2009; 187(6): 761–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Finkelstein A, Kunis G, Seksenyan A, et al.:
Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS.
PLoS One.
2011; 6(8): e22374. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Nodera H, Takamatsu N, Muguruma N, et al.:
Frequent hepatic steatosis in amyotrophic lateral sclerosis: Implication for systemic involvement.
Neurol Clin Neurosci.
2015; 3(2): 58–62. Publisher Full Text
- 23.
Bennett EJ, Mead RJ, Azzouz M, et al.:
Early detection of motor dysfunction in the SOD1G93A mouse model of Amyotrophic Lateral Sclerosis (ALS) using home cage running wheels.
PLoS One.
2014; 9(9): e107918. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Frey D, Schneider C, Xu L, et al.:
Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases.
J Neurosci.
2000; 20(7): 2534–42. PubMed Abstract
- 25.
Dobrowolny G, Aucello M, Rizzuto E, et al.:
Skeletal muscle is a primary target of SOD1G93A-mediated toxicity.
Cell Metab.
2008; 8(5): 425–36. PubMed Abstract
| Publisher Full Text
- 26.
Tsao TS, Li J, Chang KS, et al.:
Metabolic adaptations in skeletal muscle overexpressing GLUT4: effects on muscle and physical activity.
FASEB J.
2001; 15(6): 958–69. PubMed Abstract
| Publisher Full Text
- 27.
Derave W, Van Den Bosch L, Lemmens G, et al.:
Skeletal muscle properties in a transgenic mouse model for amyotrophic lateral sclerosis: effects of creatine treatment.
Neurobiol Dis.
2003; 13(3): 264–72. PubMed Abstract
| Publisher Full Text
- 28.
Toivonen JM, Manzano R, Oliván S, et al.:
MicroRNA-206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis.
PLoS One.
2014; 9(2): e89065. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Fogarty MJ, Noakes PG, Bellingham MC:
Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis.
J Neurosci.
2015; 35(2): 643–7. PubMed Abstract
| Publisher Full Text
- 30.
Graham JM, Papadakis N, Evans J, et al.:
Diffusion tensor imaging for the assessment of upper motor neuron integrity in ALS.
Neurology.
2004; 63(11): 2111–9. PubMed Abstract
| Publisher Full Text
- 31.
Ravits JM, La Spada AR:
ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration.
Neurology.
2009; 73(10): 805–811. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Nakano Y, Hirayama K, Terao K:
Hepatic ultrastructural changes and liver dysfunction in amyotrophic lateral sclerosis.
Arch Neurol.
1987; 44(1): 103–6. PubMed Abstract
| Publisher Full Text
- 33.
Fisman M:
Hepatic ultrastructural change and liver dysfunction in amyotrophic lateral sclerosis.
Arch Neurol.
1987; 44(10): 997. PubMed Abstract
| Publisher Full Text
- 34.
Zolkipli Z, Sherlock M, Biggar WD, et al.:
Abnormal fatty acid metabolism in spinal muscular atrophy may predispose to perioperative risks.
Eur J Paediatr Neurol.
2012; 16(5): 549–53. PubMed Abstract
| Publisher Full Text
- 35.
Shababi M, Lorson CL, Rudnik-Schoneborn SS:
Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease?
J Anat.
2014; 224(1): 15–28. PubMed Abstract
| Publisher Full Text
- 36.
Fusco A, Miele L, D'Uonnolo A, et al.:
Nonalcoholic fatty liver disease is associated with increased GHBP and reduced GH/IGF-I levels.
Clin Endocrinol (Oxf).
2012; 77(4): 531–6. PubMed Abstract
| Publisher Full Text
- 37.
Zoccolella S, Bendotti C, Beghi E, et al.:
Homocysteine levels and amyotrophic lateral sclerosis: A possible link.
Amyotroph Lateral Scler.
2010; 11(1–2): 140–7. PubMed Abstract
| Publisher Full Text
- 38.
DiBello PM, Dayal S, Kaveti S, et al.:
The nutrigenetics of hyperhomocysteinemia: quantitative proteomics reveals differences in the methionine cycle enzymes of gene-induced versus diet-induced hyperhomocysteinemia.
Mol Cell Proteomics.
2010; 9(3): 471–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Merriman RB:
Nonalcoholic fatty liver disease and HIV infection.
Curr HIV/AIDS Rep.
2006; 3(3): 113–7. PubMed Abstract
| Publisher Full Text
- 40.
Zein CO:
Clearing the smoke in chronic liver diseases.
Hepatology.
2010; 51(5): 1487–1490. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 41.
Agrawal P, Pandey A, Sompura S, et al.:
A rare case report showing direct association between hepatitis B and bulbar palsy.
J Assoc Physicians India.
2014; 62(3): 267–8. PubMed Abstract
- 42.
Hino H, Kusuhara T, Kaji M, et al.:
Significance of hepatitis B virus antibody in motor neuron disease.
Rinsho Shinkeigaku.
1995; 35(4): 341–3. PubMed Abstract
- 43.
Li H, Zhu W, Zhang L, et al.:
The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment.
Sci Rep.
2015; 5: 8421. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
Gupta G, Qin H, Song J:
Intrinsically unstructured domain 3 of hepatitis C Virus NS5A forms a "fuzzy complex" with VAPB-MSP domain which carries ALS-causing mutations.
PLoS One.
2012; 7(6): e39261. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 45.
Fargion S, Mattioli M, Fracanzani AL, et al.:
Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis.
Am J Gastroenterol.
2001; 96(8): 2448–55. PubMed Abstract
| Publisher Full Text
- 46.
Veyrat-Durebex C, Corcia P, Mucha A, et al.:
Iron metabolism disturbance in a French cohort of ALS patients.
BioMed Research International.
2014; 2014: 485723. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Slowik A, Tomik B, Wolkow PP, et al.:
Paraoxonase gene polymorphisms and sporadic ALS.
Neurology.
2006; 67(5): 766–70. PubMed Abstract
| Publisher Full Text
- 48.
Garcia-Heredia A, Kensicki E, Mohney RP, et al.:
Paraoxonase-1 deficiency is associated with severe liver steatosis in mice fed a high-fat high-cholesterol diet: a metabolomic approach.
J Proteome Res.
2013; 12(4): 1946–55. PubMed Abstract
| Publisher Full Text
- 49.
Li T, Owsley E, Matozel M, et al.:
Transgenic expression of cholesterol 7alpha-hydroxylase in the liver prevents high-fat diet-induced obesity and insulin resistance in mice.
Hepatology.
2010; 52(2): 678–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Dai D, Mills PB, Footitt E, et al.:
Liver disease in infancy caused by oxysterol 7 α-hydroxylase deficiency: successful treatment with chenodeoxycholic acid.
J Inherit Metab Dis.
2014; 37(5): 851–61. PubMed Abstract
| Publisher Full Text
- 51.
Tsaousidou MK, Ouahchi K, Warner TT, et al.:
Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration.
Am J Hum Genet.
2008; 82(2): 510–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 52.
Fittschen M, Lastres-Becker I, Halbach MV, et al.:
Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate.
Neurogenetics.
2015. PubMed Abstract
| Publisher Full Text
- 53.
Li P, Ruan X, Yang L, et al.:
A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice.
Cell Metab.
2015; 21(3): 455–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 54.
van den Heuvel DM, Harschnitz O, van den Berg LH, et al.:
Taking a risk: a therapeutic focus on ataxin-2 in amyotrophic lateral sclerosis?
Trends Mol Med.
2014; 20(1): 25–35. PubMed Abstract
| Publisher Full Text
- 55.
Onodera O, Akihiro S, Takuya K, et al.:
What is the key player in TDP-43 pathology in ALS: Disappearance from the nucleus or inclusion formation in the cytoplasm?
Neurology and Clinical Neuroscience.
2013; 1(1): 11–17. Publisher Full Text
- 56.
Su Q, Baker C, Christian P, et al.:
Hepatic mitochondrial and ER stress induced by defective PPARα signaling in the pathogenesis of hepatic steatosis.
Am J Physiol Endocrinol Metab.
2014; 306(11): E1264–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 57.
Pereira C:
Crosstalk between Endoplasmic Reticulum Stress and Protein Misfolding in Neurodegenerative Diseases.
ISRN Cell Biology.
2013; 2013: 22. Publisher Full Text
- 58.
Vollrath JT, Sechi A, Dreser A, et al.:
Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances.
Cell Death Dis.
2014; 5: e1290. PubMed Abstract
| Publisher Full Text
- 59.
Moriwaka F, Tashiro K, Shima K, et al.:
Glucagon and ALS.
Neurology.
1993; 43(5): 1061. PubMed Abstract
- 60.
Dodge JC, Treleaven CM, Fidler JA, et al.:
Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis.
Proc Natl Acad Sci U S A.
2013; 110(26): 10812–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Goto F, Kitamura A, Koto A, et al.:
Abnormal insulin secretion in amyotrophic lateral sclerosis.
J Neurol Sci.
1972; 16(2): 201–7. PubMed Abstract
| Publisher Full Text
- 62.
Pradat PF, Bruneteau G, Gordon PH, et al.:
Impaired glucose tolerance in patients with amyotrophic lateral sclerosis.
Amyotroph Lateral Scler.
2010; 11(1–2): 166–71. PubMed Abstract
| Publisher Full Text
- 63.
Nassir F, Ibdah JA:
Role of mitochondria in nonalcoholic fatty liver disease.
Int J Mol Sci.
2014; 15(5): 8713–8742. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Ikeda K, Hirayama T, Takazawa T, et al.:
Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study.
Intern Med.
2012; 51(12): 1501–8. PubMed Abstract
| Publisher Full Text
- 65.
den Boer M, Voshol PJ, Kuipers F, et al.:
Hepatic steatosis: A mediator of the metabolic syndrome. Lessons from animal models.
Arterioscler Thromb Vasc Biol.
2004; 24(4): 644–649. PubMed Abstract
| Publisher Full Text
- 66.
Akasaki Y, Ouchi N, Izumiya Y, et al.:
Glycolytic fast-twitch muscle fiber restoration counters adverse age-related changes in body composition and metabolism.
Aging Cell.
2014; 13(1): 80–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Li Y, Chigurupati S, Holloway HW, et al.:
Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis.
PLoS One.
2012; 7(2): e32008. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Tanaka K, Masaki Y, Tanaka M, et al.:
Exenatide improves hepatic steatosis by enhancing lipid use in adipose tissue in nondiabetic rats.
World J Gastroenterol.
2014; 20(10): 2653–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 69.
Dupuis L, Loeffler JP:
Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models.
Curr Opin Pharmacol.
2009; 9(3): 341–6. PubMed Abstract
| Publisher Full Text
- 70.
Dupuis L, Oudart H, René F, et al.:
Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model.
Proc Natl Acad Sci U S A.
2004; 101(30): 11159–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Dupuis L, di Scala F, Rene F, et al.:
Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis.
FASEB J.
2003; 17(14): 2091–3. PubMed Abstract
| Publisher Full Text
- 72.
Bernardini C, Censi F, Lattanzi W, et al.:
Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients.
PLoS One.
2013; 8(2): e57739. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 73.
Brockington A, Heath PR, Holden H, et al.:
Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis.
BMC Genomics.
2010; 11: 203. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Mali Y, Zisapels N:
Gain of interaction of ALS-linked G93A superoxide dismutase with cytosolic malate dehydrogenase.
Neurobiol Dis.
2008; 32(1): 133–41. PubMed Abstract
| Publisher Full Text
- 75.
Dunckley T, Huentelman MJ, Craig DW, et al.:
Whole-genome analysis of sporadic amyotrophic lateral sclerosis.
N Engl J Med.
2007; 357(8): 775–88. PubMed Abstract
| Publisher Full Text
- 76.
Rose AJ, Richter EA:
Skeletal muscle glucose uptake during exercise: how is it regulated?
Physiology (Bethesda).
2005; 20: 260–270. PubMed Abstract
| Publisher Full Text
- 77.
Smittkamp SE, Morris JK, Bomhoff GL, et al.:
SOD1-G93A mice exhibit muscle-fiber-type-specific decreases in glucose uptake in the absence of whole-body changes in metabolism.
Neurodegener Dis.
2014; 13(1): 29–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Perera ND, Sheean RK, Scott JW, et al.:
Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models.
PLoS One.
2014; 9(3): e90449. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Gumà A, Martínez-Redondo V, López-Soldado I, et al.:
Emerging role of neuregulin as a modulator of muscle metabolism.
Am J Physiol Endocrinol Metab.
2010; 298(4): E742–E750. PubMed Abstract
| Publisher Full Text
- 80.
Coughlan KS, Mitchem MR, Hogg MC, et al.:
“Preconditioning” with latrepirdine, an adenosine 5'-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1(G93A) mice.
Neurobiol Aging.
2015; 36(2): 1140–50. PubMed Abstract
| Publisher Full Text
- 81.
Takahashi Y, Fukuda Y, Yoshimura J, et al.:
ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19.
Am J Hum Genet.
2013; 93(5): 900–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 82.
Shaw PJ:
Motor neurone disease.
BMJ.
1999; 318(7191): 1118–1121. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Porter JD, Khanna S, Kaminski HJ, et al.:
Extraocular muscle is defined by a fundamentally distinct gene expression profile.
Proc Natl Acad Sci U S A.
2001; 98(21): 12062–12067. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 84.
Broberg S, Sahlin K:
Adenine nucleotide degradation in human skeletal muscle during prolonged exercise.
J Appl Physiol (1985).
1989; 67(1): 116–22. PubMed Abstract
- 85.
Meyer RA, Terjung RL:
Differences in ammonia and adenylate metabolism in contracting fast and slow muscle.
Am J Physiol.
1979; 237(3): C111–8. PubMed Abstract
- 86.
Phillips SC:
The toxicity to rat cerebral cortex or topical applications of acetaldehyde, ammonia or bilirubin.
Neuropathol Appl Neurobiol.
1981; 7(3): 205–16. PubMed Abstract
| Publisher Full Text
- 87.
Clay AS, Hainline BE:
Hyperammonemia in the ICU.
Chest.
2007; 132(4): 1368–1378. PubMed Abstract
| Publisher Full Text
- 88.
Walker V:
Severe hyperammonaemia in adults not explained by liver disease.
Ann Clin Biochem.
2012; 49(Pt 3): 214–28. PubMed Abstract
| Publisher Full Text
- 89.
Seiler N:
Ammonia and Alzheimer's disease.
Neurochem Int.
2002; 41(2–3): 189–207. PubMed Abstract
| Publisher Full Text
- 90.
Chiang MC, Chen HM, Lee YH, et al.:
Dysregulation of C/EBPalpha by mutant Huntingtin causes the urea cycle deficiency in Huntington's disease.
Hum Mol Genet.
2007; 16(5): 483–98. PubMed Abstract
| Publisher Full Text
- 91.
Thomsen KL, Grønbæk H, Glavind E, et al.:
Experimental nonalcoholic steatohepatitis compromises ureagenesis, an essential hepatic metabolic function.
Am J Physiol Gastrointest Liver Physiol.
2014; 307(3): G295–301. PubMed Abstract
| Publisher Full Text
- 92.
Eichler M:
Psychological changes associated with induced hyperammonemia.
Science.
1964; 144(3620): 886–8. PubMed Abstract
| Publisher Full Text
- 93.
Nardone R, Buratti T, Oliviero A, et al.:
Corticospinal involvement in patients with a portosystemic shunt due to liver cirrhosis: a MEP study.
J Neurol.
2006; 253(1): 81–5. PubMed Abstract
| Publisher Full Text
- 94.
Nardone R, Höller Y, Storti M, et al.:
Spinal cord involvement in patients with cirrhosis.
World J Gastroenterol.
2014; 20(10): 2578–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Giangaspero F, Dondi C, Scarani P, et al.:
Degeneration of the corticospinal tract following portosystemic shunt associated with spinal cord infarction.
Virchows Arch A Pathol Anat Histopathol.
1985; 406(4): 475–81. PubMed Abstract
| Publisher Full Text
- 96.
Lee KS, Kelly DL Jr:
Amyotrophic lateral sclerosis and severe cervical spondylotic myelopathy in a patient with a posterior fossa arachnoid cyst: diagnostic dilemma.
South Med J.
1987; 80(12): 1580–3. PubMed Abstract
- 97.
Braissant O, McLin VA, Cudalbu C:
Ammonia toxicity to the brain.
J Inherit Metab Dis.
2013; 36(4): 595–612. PubMed Abstract
| Publisher Full Text
- 98.
Patten BM, Kurlander HM, Evans B:
Free amino acid concentrations in spinal tissue from patients dying of motor neuron disease.
Acta Neurol Scand.
1982; 66(5): 594–9. PubMed Abstract
| Publisher Full Text
- 99.
Vanacore N, Binazzi A, Bottazzi M, et al.:
Amyotrophic lateral sclerosis in an Italian professional soccer player.
Parkinsonism Relat Disord.
2006; 12(5): 327–9. PubMed Abstract
| Publisher Full Text
- 100.
Corbett AJ, Griggs RC, Moxley RT 3rd:
Skeletal muscle catabolism in amyotrophic lateral sclerosis and chronic spinal muscular atrophy.
Neurology.
1982; 32(5): 550–2. PubMed Abstract
- 101.
Kabadi UM, Eisenstein AB, Konda J:
Elevated plasma ammonia level in hepatic cirrhosis: role of glucagon.
Gastroenterology.
1985; 88(3): 750–6. PubMed Abstract
- 102.
Hubbard RW, Will AD, Peterson GW, et al.:
Elevated plasma glucagon in amyotrophic lateral sclerosis.
Neurology.
1992; 42(8): 1532–4. PubMed Abstract
| Publisher Full Text
- 103.
Brouns F, Beckers E, Wagenmakers AJ, et al.:
Ammonia accumulation during highly intensive long-lasting cycling: individual observations.
Int J Sports Med.
1990; 11(Suppl 2): S78–84. PubMed Abstract
| Publisher Full Text
- 104.
Tomomura M, Imamura Y, Horiuchi M, et al.:
Abnormal expression of urea cycle enzyme genes in juvenile visceral steatosis (jvs) mice.
Biochim Biophys Acta.
1992; 1138(2): 167–171. PubMed Abstract
| Publisher Full Text
- 105.
Ilzecka J, Stelmasiak Z, Solski J, et al.:
Plasma amino acids concentration in amyotrophic lateral sclerosis patients.
Amino Acids.
2003; 25(1): 69–73. PubMed Abstract
| Publisher Full Text
- 106.
Nissim I, Cattano C, Lin Z, et al.:
Acid-base regulation of hepatic glutamine metabolism and ureagenesis: study with 15N.
J Am Soc Nephrol.
1993; 3(7): 1416–27. PubMed Abstract
- 107.
Bos IWM, Hoogland G, Meine Jansen CF, et al.:
Increased glutamine synthetase but normal EAAT2 expression in platelets of ALS patients.
Neurochem Int.
2006; 48(4): 306–311. PubMed Abstract
| Publisher Full Text
- 108.
Duarte-Rojo A, Torres-Vega MA, Villamil-Ramírez H, et al.:
Changes in peripheral blood mononuclear cells glutamine synthetase mRNA after exercise in healthy volunteers: exploring an alternative proposal for non hepatic ammonia metabolism.
Rev Invest Clin.
2012; 64(2): 164–72. PubMed Abstract
- 109.
Bame M, Grier RE, Needleman R, et al.:
Amino acids as biomarkers in the SOD1(G93A) mouse model of ALS.
Biochim Biophys Acta.
2014; 1842(1): 79–87. PubMed Abstract
| Publisher Full Text
- 110.
Gonzalez de Aguilar JL, Niederhauser-Wiederkehr C, Halter B, et al.:
Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model.
Physiol Genomics.
2008; 32(2): 207–18. PubMed Abstract
| Publisher Full Text
- 111.
Gonzalez de Aguilar JL, Gordon JW, René F, et al.:
A mouse model of familial amyotrophic lateral sclerosis expressing a mutant superoxide dismutase 1 shows evidence of disordered transport in the vasopressin hypothalamo-neurohypophysial axis.
Eur J Neurosci.
1999; 11(12): 4179–87. PubMed Abstract
| Publisher Full Text
- 112.
Hiroyama M, Aoyagi T, Fujiwara Y, et al.:
Hyperammonaemia in V1a vasopressin receptor knockout mice caused by the promoted proteolysis and reduced intrahepatic blood volume.
J Physiol.
2007; 581(Pt 3): 1183–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 113.
Bobermin LD, Quincozes-Santos A, Guerra MC, et al.:
Resveratrol prevents ammonia toxicity in astroglial cells.
PLoS One.
2012; 7(12): e52164. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 114.
Smith W, Diaz GA, Lichter-Konecki U, et al.:
Ammonia control in children ages 2 months through 5 years with urea cycle disorders: comparison of sodium phenylbutyrate and glycerol phenylbutyrate.
J Pediatr.
2013; 162(6): 1228–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 115.
Lee J, Ryu H, Kowall NW:
Motor neuronal protection by L-arginine prolongs survival of mutant SOD1 (G93A) ALS mice.
Biochem Biophys Res Commun.
2009; 384(4): 524–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 116.
Mizutani N, Kato T, Maehara M, et al.:
Oral administration of arginine and citrulline in the treatment of lysinuric protein intolerance.
Tohoku J Exp Med.
1984; 142(1): 15–24. PubMed Abstract
| Publisher Full Text
- 117.
Kira Y, Nishikawa M, Ochi A, et al.:
L-carnitine suppresses the onset of neuromuscular degeneration and increases the life span of mice with familial amyotrophic lateral sclerosis.
Brain Res.
2006; 1070(1): 206–14. PubMed Abstract
| Publisher Full Text
- 118.
Mancuso R, del Valle J, Modol L, et al.:
Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice.
Neurotherapeutics.
2014; 11(2): 419–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 119.
Nunn PB, Ponnusamy M:
Beta-N-methylaminoalanine (BMAA): metabolism and metabolic effects in model systems and in neural and other tissues of the rat in vitro.
Toxicon.
2009; 54(2): 85–94. PubMed Abstract
| Publisher Full Text
- 120.
de Munck E, Muñoz-Sáez E, Antonio MT, et al.:
Effect of β-N-methylamino-L-alanine on oxidative stress of liver and kidney in rat.
Environ Toxicol Pharmacol.
2013; 35(2): 193–9. PubMed Abstract
| Publisher Full Text
- 121.
O'Neal RM, Chen CH, Reynolds CS, et al.:
The 'neurotoxicity' of L-2,4-diaminobutyric acid.
Biochem J.
1968; 106(3): 699–706. PubMed Abstract
| Free Full Text
- 122.
Cheema PS, Malathi K, Padmanaban G, et al.:
The neurotoxicity of beta-N-oxalyl-L-alphabeta-diaminopropionic acid, the neurotoxin from the pulse Lathyrus sativus.
Biochem J.
1969; 112(1): 29–33. PubMed Abstract
| Free Full Text
- 123.
Doi H, Kikuchi H, Murai H, et al.:
Motor neuron disorder simulating ALS induced by chronic inhalation of pyrethroid insecticides.
Neurology.
2006; 67(10): 1894–5. PubMed Abstract
| Publisher Full Text
- 124.
Kumar A, Sharma B, Pandey RS:
Cypermethrin induced alterations in nitrogen metabolism in freshwater fishes.
Chemosphere.
2011; 83(4): 492–501. PubMed Abstract
| Publisher Full Text
- 125.
Tada M, Coon EA, Osmand AP, et al.:
Coexistence of Huntington's disease and amyotrophic lateral sclerosis: a clinicopathologic study.
Acta Neuropathol.
2012; 124(5): 749–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 126.
Sadeghian H, O'Suilleabhain PE, Battiste J, et al.:
Huntington chorea presenting with motor neuron disease.
Arch Neurol.
2011; 68(5): 650–2. PubMed Abstract
| Publisher Full Text
- 127.
Chiang MC, Chern Y, Juo CG:
The dysfunction of hepatic transcriptional factors in mice with Huntington's Disease.
Biochim Biophys Acta.
2011; 1812(9): 1111–1120. PubMed Abstract
| Publisher Full Text
- 128.
Hensman Moss DJ, Poulter M, Beck J, et al.:
C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies.
Neurology.
2014; 82(4): 292–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 129.
Berger R, Mezey E, Clancy KP, et al.:
Analysis of aldehyde oxidase and xanthine dehydrogenase/oxidase as possible candidate genes for autosomal recessive familial amyotrophic lateral sclerosis.
Somat Cell Mol Genet.
1995; 21(2): 121–31. PubMed Abstract
| Publisher Full Text
- 130.
Ekblom J, Aquilonius SM, Jossan SS:
Differential increases in catecholamine metabolizing enzymes in amyotrophic lateral sclerosis.
Exp Neurol.
1993; 123(2): 289–94. PubMed Abstract
| Publisher Full Text
- 131.
Orru S, Mascia V, Casula M, et al.:
Association of monoamine oxidase B alleles with age at onset in amyotrophic lateral sclerosis.
Neuromuscul Disord.
1999; 9(8): 593–7. PubMed Abstract
| Publisher Full Text
- 132.
Skowronska M, Albrecht J:
Oxidative and nitrosative stress in ammonia neurotoxicity.
Neurochem Int.
2013; 62(5): 731–7. PubMed Abstract
| Publisher Full Text
- 133.
Butterworth RF:
Glutamate transporters in hyperammonemia.
Neurochem Int.
2002; 41(2–3): 81–85. PubMed Abstract
| Publisher Full Text
- 134.
Tsuboi M, Harasawa K, Izawa T, et al.:
Intralysosomal pH and release of lysosomal enzymes in the rat liver after exhaustive exercise.
J Appl Physiol (1985).
1993; 74(4): 1628–34. PubMed Abstract
- 135.
Cagnon L, Braissant O:
Role of caspases, calpain and cdk5 in ammonia-induced cell death in developing brain cells.
Neurobiol Dis.
2008; 32(2): 281–92. PubMed Abstract
| Publisher Full Text
- 136.
Gorg B, Karababa A, Shafigullina A, et al.:
Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy.
Glia.
2015; 63(1): 37–50. PubMed Abstract
| Publisher Full Text
- 137.
Yamanaka K, Chun SJ, Boillee S, et al.:
Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis.
Nat Neurosci.
2008; 11(3): 251–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Nicaise C, Mitrecic D, Demetter P, et al.:
Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat.
Brain Res.
2009; 1301: 152–62. PubMed Abstract
| Publisher Full Text
- 139.
Chen Y, Stankovic R, Cullen KM, et al.:
The kynurenine pathway and inflammation in amyotrophic lateral sclerosis.
Neurotox Res.
2010; 18(2): 132–42. PubMed Abstract
| Publisher Full Text
- 140.
Okamoto K, Hirai S, Amari M, et al.:
Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum.
Neurosci Lett.
1993; 162(1–2): 125–8. PubMed Abstract
| Publisher Full Text
- 141.
van Dis V, Kuijpers M, Haasdijk ED, et al.:
Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons.
Acta Neuropathol Commun.
2014; 2: 38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 142.
Kundu SK, Harati Y, Misra LK:
Sialosylglobotetraosylceramide: a marker for amyotropic lateral sclerosis.
Biochem Biophys Res Commun.
1984; 118(1): 82–9. PubMed Abstract
- 143.
Bajaj NP:
Cyclin-dependent kinase-5 (CDK5) and amyotrophic lateral sclerosis.
Amyotroph Lateral Scler Other Motor Neuron Disord.
2000; 1(5): 319–27. PubMed Abstract
| Publisher Full Text
- 144.
Okamoto K, Hirai S, Shoji M, et al.:
Axonal swellings in the corticospinal tracts in amyotrophic lateral sclerosis.
Acta Neuropathol.
1990; 80(2): 222–6. PubMed Abstract
| Publisher Full Text
- 145.
Kihira T, Mizusawa H, Tada J, et al.:
Lewy body-like inclusions in Onuf's nucleus from two cases of sporadic amyotrophic lateral sclerosis.
J Neurol Sci.
1993; 115(1): 51–7. PubMed Abstract
| Publisher Full Text
- 146.
Sahenk Z, Brown A:
Weak-base amines inhibit the anterograde-to-retrograde conversion of axonally transported vesicles in nerve terminals.
J Neurocytol.
1991; 20(5): 365–375. PubMed Abstract
| Publisher Full Text
- 147.
Dean RT, Jessup W, Roberts CR:
Effects of exogenous amines on mammalian cells, with particular reference to membrane flow.
Biochem J.
1984; 217(1): 27–40. PubMed Abstract
| Free Full Text
- 148.
Shacka JJ, Klocke BJ, Young C, et al.:
Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis.
J Neurosci.
2007; 27(8): 2081–90. PubMed Abstract
| Publisher Full Text
- 149.
Chen S, Zhang X, Song L, et al.:
Autophagy dysregulation in amyotrophic lateral sclerosis.
Brain Pathol.
2012; 22(1): 110–6. PubMed Abstract
| Publisher Full Text
- 150.
Rubinsztein DC, Ravikumar B, Acevedo-Arozena A, et al.:
Dyneins, autophagy, aggregation and neurodegeneration.
Autophagy.
2005; 1(3): 177–8. PubMed Abstract
| Publisher Full Text
- 151.
Hafezparast M, Klocke R, Ruhrberg C, et al.:
Mutations in dynein link motor neuron degeneration to defects in retrograde transport.
Science.
2003; 300(5620): 808–12. PubMed Abstract
| Publisher Full Text
- 152.
Wilson ME, Boumaza I, Bowser R:
Measurement of cystatin C functional activity in the cerebrospinal fluid of amyotrophic lateral sclerosis and control subjects.
Fluids Barriers CNS.
2013; 10(1): 15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 153.
Yates CM, Wilson H, Davidson D:
Lysosomal enzymes in motor neurone disease and multiple sclerosis.
Clin Chim Acta.
1973; 47(3): 397–402. PubMed Abstract
| Publisher Full Text
- 154.
Nagy L, Kusstatscher S, Hauschka PV, et al.:
Role of cysteine proteases and protease inhibitors in gastric mucosal damage induced by ethanol or ammonia in the rat.
J Clin Invest.
1996; 98(4): 1047–1054. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 155.
Wootz H, Weber E, Korhonen L, et al.:
Altered distribution and levels of cathepsinD and cystatins in amyotrophic lateral sclerosis transgenic mice: possible roles in motor neuron survival.
Neuroscience.
2006; 143(2): 419–30. PubMed Abstract
| Publisher Full Text
- 156.
Kikuchi H, Yamada T, Furuya H, et al.:
Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis.
Acta Neuropathol.
2003; 105(5): 462–8. PubMed Abstract
| Publisher Full Text
- 157.
Lee S, Sato Y, Nixon RA:
Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy.
J Neurosci.
2011; 31(21): 7817–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 158.
Bryson JB, Hobbs C, Parsons MJ, et al.:
Amyloid precursor protein (APP) contributes to pathology in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis.
Hum Mol Genet.
2012; 21(17): 3871–82. PubMed Abstract
| Publisher Full Text
- 159.
Guo Y, Li C, Wu D, et al.:
Ultrastructural diversity of inclusions and aggregations in the lumbar spinal cord of SOD1-G93A transgenic mice.
Brain Res.
2010; 1353: 234–244. PubMed Abstract
| Publisher Full Text
- 160.
Xiao S, McLean J, Robertson J:
Neuronal intermediate filaments and ALS: A new look at an old question.
Biochim Biophys Acta.
2006; 1762(11–12): 1001–1012. PubMed Abstract
| Publisher Full Text
- 161.
Letournel F, Bocquet A, Dubas F, et al.:
Stable tubule only polypeptides (STOP) proteins co-aggregate with spheroid neurofilaments in amyotrophic lateral sclerosis.
J Neuropathol Exp Neurol.
2003; 62(12): 1211–9. PubMed Abstract
- 162.
McHolm GB, Aguilar MJ, Norris FH:
Lipofuscin in amyotrophic lateral sclerosis.
Arch Neurol.
1984; 41(11): 1187–8. PubMed Abstract
| Publisher Full Text
- 163.
Yang EJ, Choi SM:
α -Synuclein Modification in an ALS Animal Model.
Evid Based Complement Alternat Med.
2013; 2013: 259381. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 164.
Crabtree D, Dodson M, Ouyang X, et al.:
Over-expression of an inactive mutant cathepsin D increases endogenous alpha-synuclein and cathepsin B activity in SH-SY5Y cells.
J Neurochem.
2014; 128(6): 950–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 165.
Asai M, Yagishita S, Iwata N, et al.:
An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model.
FASEB J.
2011; 25(10): 3720–30. PubMed Abstract
| Publisher Full Text
- 166.
Nakano I, Shibata T, Uesaka Y:
On the possibility of autolysosomal processing of skein-like inclusions. Electron microscopic observation in a case of amyotrophic lateral sclerosis.
J Neurol Sci.
1993; 120(1): 54–9. PubMed Abstract
| Publisher Full Text
- 167.
Korolchuk VI, Menzies FM, Rubinsztein DC:
Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems.
FEBS Lett.
2010; 584(7): 1393–1398. PubMed Abstract
| Publisher Full Text
- 168.
Sonnino S, Chigorno V:
Ganglioside molecular species containing C18- and C20-sphingosine in mammalian nervous tissues and neuronal cell cultures.
Biochim Biophys Acta.
2000; 1469(2): 63–77. PubMed Abstract
| Publisher Full Text
- 169.
Matsumoto A, Yoshino H, Yuki N, et al.:
Ganglioside characterization of a cell line displaying motor neuron-like phenotype: GM2 as a possible major ganglioside in motor neurons.
J Neurol Sci.
1995; 131(2): 111–8. PubMed Abstract
| Publisher Full Text
- 170.
Johnson WG:
Motor neuron diseases resulting from hexosaminidase deficiency.
Semin Neurol.
1993; 13(4): 369–74. PubMed Abstract
| Publisher Full Text
- 171.
Johnson WG:
Hexosaminidase deficiency: a cause of recessively inherited motor neuron diseases.
Adv Neurol.
1982; 36: 159–64. PubMed Abstract
- 172.
Banerjee P, Siciliano L, Oliveri D, et al.:
Molecular basis of an adult form of beta-hexosaminidase B deficiency with motor neuron disease.
Biochem Biophys Res Commun.
1991; 181(1): 108–15. PubMed Abstract
| Publisher Full Text
- 173.
Dawson G, Stefansson K:
Gangliosides of human spinal cord: aberrant composition of cords from patients with amyotrophic lateral sclerosis.
J Neurosci Res.
1984; 12(2–3): 213–20. PubMed Abstract
| Publisher Full Text
- 174.
Rapport MM, Donnenfeld H, Brunner W, et al.:
Ganglioside patterns in amyotrophic lateral sclerosis brain regions.
Ann Neurol.
1985; 18(1): 60–67. PubMed Abstract
| Publisher Full Text
- 175.
Modi P, Sadasivudu B, Lakshminarayana U, et al.:
Functional relationship between ammonia and gangliosides in brain.
Neurochem Res.
1994; 19(3): 353–8. PubMed Abstract
| Publisher Full Text
- 176.
Perez LF, Casal JA, Rojas P, et al.:
Relationship between plasma ammonia concentration and β-N-acetylhexosaminidase isoenzyme activities in liver cirrhosis.
Clin Chem Lab Med.
2000; 38(12): 1237–41. PubMed Abstract
| Publisher Full Text
- 177.
Segers K, Kadhim H, Colson C, et al.:
Adult polyglucosan body disease masquerading as “ALS with dementia of the Alzheimer type”: an exceptional phenotype in a rare pathology.
Alzheimer Dis Assoc Disord.
2012; 26(1): 96–9. PubMed Abstract
| Publisher Full Text
- 178.
Robitaille Y, Carpenter S, Karpati G, et al.:
A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora's disease and normal ageing.
Brain.
1980; 103(2): 315–36. PubMed Abstract
| Publisher Full Text
- 179.
Atanassov CL, Muller CD, Sarhan S, et al.:
Effect of ammonia on endocytosis, cytokine production and lysosomal enzyme activity of a microglial cell line.
Res Immunol.
1994; 145(4): 277–88. PubMed Abstract
| Publisher Full Text
- 180.
McDonald TD, Faust PL, Bruno C, et al.:
Polyglucosan body disease simulating amyotrophic lateral sclerosis.
Neurology.
1993; 43(4): 785–90. PubMed Abstract
| Publisher Full Text
- 181.
Sun KH, de Pablo Y, Vincent F, et al.:
Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction.
J Neurochem.
2008; 107(1): 265–78. PubMed Abstract
| Publisher Full Text
- 182.
Nguyen MD, Boudreau M, Kriz J, et al.:
Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1.
J Neurosci.
2003; 23(6): 2131–40. PubMed Abstract
- 183.
Ralay Ranaivo H, Hodge JN, Choi N, et al.:
Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways.
J Neuroinflammation.
2012; 9: 68. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Lukaszewicz-Zajac M, Mroczko B, Slowik A:
Matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in amyotrophic lateral sclerosis (ALS).
J Neural Transm.
2014; 121(11): 1387–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 185.
Kaplan A, Spiller KJ, Towne C, et al.:
Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration.
Neuron.
2014; 81(2): 333–48. PubMed Abstract
| Publisher Full Text
- 186.
Skowronska M, Zielińska M, Wójcik-Stanaszek L, et al.:
Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases.
J Neurochem.
2012; 121(1): 125–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 187.
Reszka AA, Seger R, Diltz CD, et al.:
Association of mitogen-activated protein kinase with the microtubule cytoskeleton.
Proc Natl Acad Sci U S A
1995; 92(19): 8881–8885. PubMed Abstract
| Free Full Text
- 188.
Li JJ, Ji R, Shi YQ, et al.:
Changes in expression of the chloride homeostasis-regulating genes, KCC2 and NKCC1, in the blood of cirrhotic patients with hepatic encephalopathy.
Exp Ther Med.
2012; 4(6): 1075–1080. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 189.
Hübner CA, Stein V, Hermans-Borgmeyer I, et al.:
Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition.
Neuron.
2001; 30(2): 515–524. PubMed Abstract
| Publisher Full Text
- 190.
Fuchs A, Ringer C, Bilkei-Gorzo A, et al.:
Downregulation of the potassium chloride cotransporter KCC2 in vulnerable motoneurons in the SOD1-G93A mouse model of amyotrophic lateral sclerosis.
J Neuropathol Exp Neurol.
2010; 69(10): 1057–70. PubMed Abstract
| Publisher Full Text
- 191.
Ramirez-Jarquin UN, Lazo-Gómez R, Tovar-Y-Romo LB, et al.:
Spinal inhibitory circuits and their role in motor neuron degeneration.
Neuropharmacology.
2014; 82: 101–7. PubMed Abstract
| Publisher Full Text
- 192.
Jayakumar AR, Panickar KS, Murthy ChR, et al.:
Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes.
J Neurosci.
2006; 26(18): 4774–84. PubMed Abstract
| Publisher Full Text
- 193.
Zhou BG, Norenberg MD:
Ammonia downregulates GLAST mRNA glutamate transporter in rat astrocyte cultures.
Neurosci Lett.
1999; 276(3): 145–8. PubMed Abstract
| Publisher Full Text
- 194.
Howland DS, Liu J, She Y, et al.:
Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS).
Proc Natl Acad Sci U S A.
2002; 99(3): 1604–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 195.
Heath PR, Shaw PJ:
Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis.
Muscle Nerve.
2002; 26(4): 438–58. PubMed Abstract
| Publisher Full Text
- 196.
Dunlop J, Beal McIlvain H, She Y, et al.:
Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis.
J Neurosci.
2003; 23(5): 1688–96. PubMed Abstract
- 197.
Felipo V, Butterworth RF:
Neurobiology of ammonia.
Prog Neurobiol.
2002; 67(4): 259–79. PubMed Abstract
| Publisher Full Text
- 198.
Reinehr R, Görg B, Becker S, et al.:
Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices.
Glia.
2007; 55(7): 758–71. PubMed Abstract
| Publisher Full Text
- 199.
Jayakumar AR, Tong XY, Curtis KM, et al.:
Increased toll-like receptor 4 in cerebral endothelial cells contributes to the astrocyte swelling and brain edema in acute hepatic encephalopathy.
J Neurochem.
2014; 128(6): 890–903. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 200.
Rodrigo R, Cauli O, Gomez-Pinedo U, et al.:
Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy.
Gastroenterology.
2010; 139(2): 675–84. PubMed Abstract
| Publisher Full Text
- 201.
Guillemin GJ, Meininger V, Brew BJ:
Implications for the kynurenine pathway and quinolinic acid in amyotrophic lateral sclerosis.
Neurodegener Dis.
2005; 2(3–4): 166–76. PubMed Abstract
| Publisher Full Text
- 202.
Marrali G, Casale F, Salamone P, et al.:
NADPH oxidase (NOX2) activity is a modifier of survival in ALS.
J Neurol.
2014; 261(11): 2178–83. PubMed Abstract
| Publisher Full Text
- 203.
Casula M, Iyer AM, Spliet WG, et al.:
Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue.
Neuroscience.
2011; 179: 233–43. PubMed Abstract
| Publisher Full Text
- 204.
Evans MC, Couch Y, Sibson N, et al.:
Inflammation and neurovascular changes in amyotrophic lateral sclerosis.
Mol Cell Neurosci.
2013; 53: 34–41. PubMed Abstract
| Publisher Full Text
- 205.
Appel SH, Beers D, Siklos L, et al.:
Calcium: the Darth Vader of ALS.
Amyotroph Lateral Scler Other Motor Neuron Disord.
2001; 2(Suppl 1): S47–54. PubMed Abstract
| Publisher Full Text
- 206.
Ince P, Stout N, Shaw P, et al.:
Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease.
Neuropathol Appl Neurobiol.
1993; 19(4): 291–9. PubMed Abstract
| Publisher Full Text
- 207.
Copray JC, Liem RS, Kernell D:
Calreticulin expression in spinal motoneurons of the rat.
J Chem Neuroanat.
1996; 11(1): 57–65. PubMed Abstract
| Publisher Full Text
- 208.
Bernard-Marissal N, Sunyach C, Marissal T, et al.:
Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice.
Neurobiol Dis.
2015; 73: 130–6. PubMed Abstract
| Publisher Full Text
- 209.
Bernard-Marissal N, Moumen A, Sunyach C, et al.:
Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS.
J Neurosci.
2012; 32(14): 4901–12. PubMed Abstract
| Publisher Full Text
- 210.
Montague K, Malik B, Gray AL, et al.:
Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy.
Brain.
2014; 137(Pt 7): 1894–906. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 211.
Drake ME Jr:
The association of motor neuron disease and Alzheimer-type dementia.
Am J Med Sci.
1984; 287(3): 26–7. PubMed Abstract
| Publisher Full Text
- 212.
Crugnola V, Lamperti C, Lucchini V, et al.:
Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis.
Arch Neurol.
2010; 67(7): 849–54. PubMed Abstract
| Publisher Full Text
- 213.
Menzies FM, Ince PG, Shaw PJ:
Mitochondrial involvement in amyotrophic lateral sclerosis.
Neurochem Int.
2002; 40(6): 543–51. PubMed Abstract
| Publisher Full Text
- 214.
Carr PA, Yamamoto T, Karmy G, et al.:
Analysis of parvalbumin and calbindin D28k-immunoreactive neurons in dorsal root ganglia of rat in relation to their cytochrome oxidase and carbonic anhydrase content.
Neuroscience.
1989; 33(2): 363–71. PubMed Abstract
| Publisher Full Text
- 215.
Linnebank M, Lutz H, Jarre E, et al.:
Binding of copper is a mechanism of homocysteine toxicity leading to COX deficiency and apoptosis in primary neurons, PC12 and SHSY-5Y cells.
Neurobiol Dis.
2006; 23(3): 725–30. PubMed Abstract
| Publisher Full Text
- 216.
Patel SP, Gamboa JL, McMullen CA, et al.:
Lower respiratory capacity in extraocular muscle mitochondria: evidence for intrinsic differences in mitochondrial composition and function.
Invest Ophthalmol Vis Sci.
2009; 50(1): 180–186. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 217.
Andrade FH, McMullen CA, Rumbaut RE:
Mitochondria are fast Ca2+ sinks in rat extraocular muscles: a novel regulatory influence on contractile function and metabolism.
Invest Ophthalmol Vis Sci.
2005; 46(12): 4541–7. PubMed Abstract
| Publisher Full Text
- 218.
Beauchemin AM, Gottlieb B, Beitel LK, et al.:
Cytochrome c oxidase subunit Vb interacts with human androgen receptor: a potential mechanism for neurotoxicity in spinobulbar muscular atrophy.
Brain Res Bull.
2001; 56(3–4): 285–97. PubMed Abstract
| Publisher Full Text
- 219.
Comi GP, Bordoni A, Salani S, et al.:
Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease.
Ann Neurol.
1998; 43(1): 110–6. PubMed Abstract
| Publisher Full Text
- 220.
Rygiel KA, Grady JP, Turnbull DM:
Respiratory chain deficiency in aged spinal motor neurons.
Neurobiol Aging.
2014; 35(10): 2230–2238. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 221.
Sullivan PG, Rabchevsky AG, Keller JN, et al.:
Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions.
J Comp Neurol.
2004; 474(4): 524–34. PubMed Abstract
| Publisher Full Text
- 222.
Turner C, Parekh B, Walton C, et al.:
An exploratory comparative study of volatile compounds in exhaled breath and emitted by skin using selected ion flow tube mass spectrometry.
Rapid Commun Mass Spectrom.
2008; 22(4): 526–32. PubMed Abstract
| Publisher Full Text
- 223.
Wright G, Noiret L, Olde Damink SW, et al.:
Interorgan ammonia metabolism in liver failure: the basis of current and future therapies.
Liver Int.
2011; 31(2): 163–75. PubMed Abstract
| Publisher Full Text
- 224.
Diaz-Herrero MM, del Campo JA, Carbonero-Aguilar P, et al.:
THDP17 decreases ammonia production through glutaminase inhibition. A new drug for hepatic encephalopathy therapy.
PLoS One.
2014; 9(10): e109787. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 225.
Kristiansen RG, Rose CF, Fuskevåg OM, et al.:
L-Ornithine phenylacetate reduces ammonia in pigs with acute liver failure through phenylacetylglycine formation: a novel ammonia-lowering pathway.
Am J Physiol Gastrointest Liver Physiol.
2014; 307(10): G1024–31. PubMed Abstract
| Publisher Full Text
- 226.
Ikarashi N, Fukazawa Y, Toda T, et al.:
Effect of Conclevan on endurance capacity in mice.
Biol Pharm Bull.
2012; 35(2): 231–8. PubMed Abstract
| Publisher Full Text
- 227.
Matthys D, Calders P, Pannier JL:
Inhaled salbutamol decreases blood ammonia levels during exercise in normal subjects.
Eur J Appl Physiol Occup Physiol.
1998; 79(1): 110–3. PubMed Abstract
| Publisher Full Text
- 228.
Gruzman A, Shamni O, Ben Yakir M, et al.:
Novel D-xylose derivatives stimulate muscle glucose uptake by activating AMP-activated protein kinase alpha.
J Med Chem.
2008; 51(24): 8096–108. PubMed Abstract
| Publisher Full Text
- 229.
Coelho WS, Costa KC, Sola-Penna M:
Serotonin stimulates mouse skeletal muscle 6-phosphofructo-1-kinase through tyrosine-phosphorylation of the enzyme altering its intracellular localization.
Mol Genet Metab.
2007; 92(4): 364–70. PubMed Abstract
| Publisher Full Text
- 230.
Rangroo Thrane V, Thrane AS, Wang F, et al.:
Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering.
Nat Med.
2013; 19(12): 1643–1648. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 231.
Osher E, Fattal-Valevski A, Sagie L, et al.:
Pyrimethamine increases beta-hexosaminidase A activity in patients with Late Onset Tay Sachs.
Mol Genet Metab.
2011; 102(3): 356–63. PubMed Abstract
| Publisher Full Text
- 232.
Kiernan MC, Vucic S, Cheah BC, et al.:
Amyotrophic lateral sclerosis.
The Lancet.
2011; 377(9769): 942–955. PubMed Abstract
| Publisher Full Text
Comments on this article Comments (0)