Keywords
Anesthesia, Niemann-Pick disease, genomic and biochemical analysis
Anesthesia, Niemann-Pick disease, genomic and biochemical analysis
Niemann-Pick disease (NPD) is a rare inherited autosomal recessive lysosomal storage disorder (incidence about 1:40,000 in general population) caused by pathogenic mutations in the SMPD1 gene and characterised by enzyme studies showing a corresponding very low level of enzymatic activity of acidic sphingomyelinase (ASM), associated with intracellular accumulation of lipids1–3. People with NPD type A (NPA; generally a very rare presentation of NPD) have little or no ASM production (less than 1% of normal) while those with NPD type B (the most frequent presentation of NPD) have approximately 10% of the normal level of ASM. There is no information on the anesthetic management of a patient with fully genetically characterized and biochemically confirmed NPA, and only one previous report describing a pediatric patient with presumably diagnosed NPA4. In this case report, we describe the genetic background, pathophysiology and anesthetic-related problems in a patient with NPA who presented for surgery.
A 14-month-old Caucasian child (residing in the United States, diagnosed with NPA, presented to our hospital pre-anesthesia assessment clinic for laparoscopic placement of a gastrostomy tube. There was no known family member or relatives diagnosed with NPD; mother reported a distant relative of Jewish origin. On physical exam, he was a well-proportioned child, weight was 8.32 kg (10th percentile) and height was 77cm (50–75th percentile), head circumference was 48.3 cm (90–95th percentile). Craniofacial features included frontal bossing, protuberant tongue and mild bilateral ptosis. Generalized hypotonia with head lag and weakness in upper girdle and lower leg muscles, decreased deep tendon reflexes and hepatosplenomegaly (3 cm below the costal margin) were noted. Liver function tests revealed elevation of alanine transaminase (ALT) levels - 90 uts/L (normal range 13–69), aspartate transaminase (AST) levels - 271 uts/L (normal range 9–80), alkaline phosphatase levels - 252 uts/L (normal 8–240) and creatine phosphokinase (CPK) - <20 uts/L (normal range 55–170).
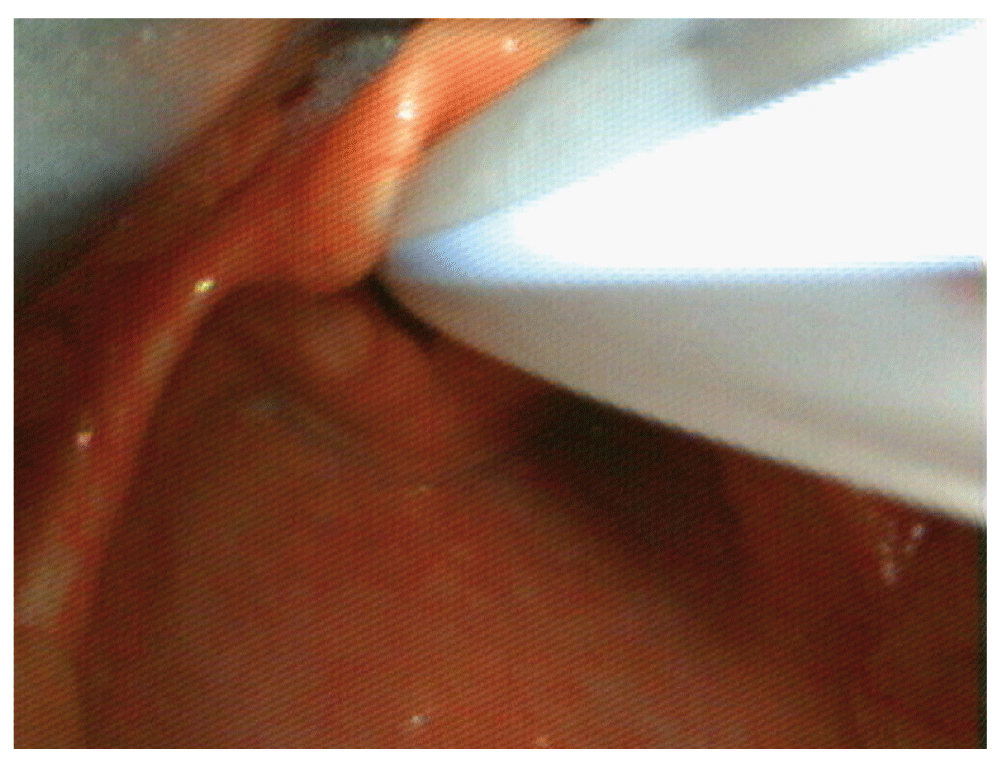
The child was scheduled as the first case on the day of surgery. General anesthesia was induced with a 50:50 mixture of nitrous oxide: oxygen and sevoflurane. Following a smooth inhalational induction, intravenous access was established and a single dose of propofol 2 mg/kg was administered intravenously to facilitate tracheal intubation. Tracheal intubation was performed under deep anesthesia with a 4 mm internal diameter cuffed endotracheal tube with a c-MAC videolaryngoscope blade (Cormack and Lehane grade 2 view, Figure 1). Anesthesia was maintained with air: oxygen and sevoflurane mixture (Minimum Alveolar Concentration i.e. MAC=1). A single dose of per rectal acetaminophen 240 mg (30–40 mg/kg) and local anesthetic infiltration with 0.25% bupivacaine were used to facilitate analgesia. Tracheal extubation was uneventful. The child was transported to the post-anesthesia care unit and made an uneventful post-operative recovery.
Follow-up The child had G-tube feeds commenced following surgery and subsequently discharged to home the following day and made an uneventful clinical recovery. Subsequently over the course of next few months, the child had episodes of intractable seizures which were medically managed. The patient was referred to home hospice care and expired at home at the age of 2 years.
The results of enzymatic evaluation of patient blood and leukocytes by an outside laboratory are presented in Table 1.
Extracted DNA was PCR-amplified for analysis of the coding exons 1 to 6 of the SPMD1 gene and their flanking splice sites, using a standard Sanger sequencing approach. Bi-directional sequence was obtained and the DNA sequence was analyzed and compared to the published gene sequence. Reportable variants were confirmed by repeat sequence analysis. Based on the genetic diagnostic laboratory (Proprietary information from GeneDx, Gaithersburg, MD) 99% sensitivity is expected in detecting mutations identifiable by sequencing. Please refer to Table 2.
Our anesthesia management strategy focused on avoiding muscle relaxants and narcotic analgesics. Hence, endotracheal intubation under deep anesthesia without muscle paralysis and non-opioid analgesics, i.e., acetaminophen, and infiltration of the surgical wound with local anesthetic were contemplated. Adequate pain control was possible with this technique since the procedure was performed laparoscopically. Although the child had mild elevation of liver enzymes, acetaminophen was preferred as a short-term analgesic. Use of atracurium, midazolam, sevoflurane and fentanyl has been described in the management of anesthesia in a 2-year-old patient with presumable NPA presenting for emergency splenectomy4. The child had severe hepatosplenomegaly, ventilation improved after splenectomy and the child required a post-operative ICU stay for 9 days.
The observed c.573delT mutation in the SMPD1 gene in our case has been reported previously in association with NPD type A5. The deletion causes a frame shift starting with codon Serine 192, changes this amino acid to an Alanine residue and creates a premature Stop codon at position 65 in the new reading frame denoted p.Ser192AlafsX65. This mutation is predicted to cause abnormal protein function either through protein truncation or nonsense-mediated mRNA decay. The c1783_1784delCT deletion causes a frame shift starting with codon Alanine 597, changes this amino acid to a Proline residue, and creates a premature stop codon at position 7 of the new reading frame denoted p.Ala597ProfsX7. This mutation is predicted to cause loss of normal protein function through protein truncation. Specifically, the last 35 correct residues are replaced by six incorrect residues. Therefore, the presence of these mutations is consistent with the diagnosis of an SMPD1-related disorder, if the mutations were inherited on different alleles (in trans), i.e., each mutation coming from a different parent6. This result therefore permits mutation-specific carrier testing for family members and prenatal diagnosis for the parents of this child, if desired. However, targeted carrier testing of both parents is necessary prior to or concurrently with any carrier testing or predictive testing in this family.
Patients with NPA and NPB have significantly different clinical course. While NPA is associated with severe neurological deficits leading to early death, patients with NPB have minimal neurological involvement and may survive to adulthood, although hepatosplenomegaly and cardiorespiratory problems may occur. We believe therefore that it is important to distinguish these form of NPD preoperatively, because NPA requires much more strict anesthetic management (avoidance of muscle relaxants and opioids, if at all possible) and it could associated more frequently with step-up level of postoperative care.
Written informed consent for publication of clinical details and/or images was obtained from the parent of the patient.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)