Keywords
Hypoxia, CoCl2, renal cancer, gene expression, metabolism pathways
Hypoxia, CoCl2, renal cancer, gene expression, metabolism pathways
Hypoxia is characterized by reduced oxygen supply and appears in multiple pathological conditions including tumours. However, hypoxia can also have a functional role during normal mammalian development and embryogenesis 1 . Cells respond to hypoxic conditions both on biochemical and gene expression levels by switching from aerobic metabolism to anaerobic glycolysis and by expression of stress-related genes involved in regulation of cell death, erythropoiesis, angiogenesis and survival 2–4 . The activation of many O2-regulated genes is mediated by hypoxia-inducible factor (Hif1a). Under normoxia, Hif1a is hydroxylated by specific prolyl hydroxylases (PHD1, PHD2 and PHD3). This reaction requires oxygen, 2-oxoglutarate and ascorbate 5,6 . When Hif1a is hydroxylated, it interacts with the von Hippel-Lindau tumor suppressor protein (pVHL). pVHL forms the substrate-recognition module of an E3 ubiquitin ligase complex, which directs Hif1a poly-ubiquitylation and proteasomal degradation 7,8 . Under hypoxia (less than 5% O2), PHD activity is inhibited by cytoplasmic reactive-oxygen species (ROS) which alter the oxidation state of Fe2+ (a cofactor for PHD activity) to Fe3+. This alteration inhibits PHD activity and Hif1a hydroxylation, thus Hif1a cannot interact with pVHL and promotes HIf1a stabilization 9,10 . This anaerobic condition and stabilization of Hif1a are characteristic of many tumors. The most common molecular abnormality in renal cell carcinoma is the loss of VHL, which is found in about 50–70% of sporadic cases. Consequently, renal carcinomas with mutations in VHL have high steady-state levels of Hif1a expression and are hypoxic 11 . Some divalent cations such as cobalt (Co2+), nickel (Ni2+), and the iron-chelator deferoxamine (DFX), have been applied to mimic hypoxic conditions in cultured cells as they activate hypoxic signals by stabilizing HIF1a 12 . Transition metal Co2+ could induce hypoxic response by inhibiting PHD activity via iron replacement. Therefore, treatment of a cell culture with cobalt chloride (CoCl2) is a common model of hypoxia 13 . The second classical setup to study hypoxia is hypoxia induction in a CO2 incubator with a regulated level of oxygen (less than 1% O2). In this work, we performed RNA sequencing of Caki-1 clear cell renal cancer cell lines treated with hypoxia and with CoCl2 to understand how adequate CoCl2 treatment was as a hypoxia model. We propose that CoCl2 is not a completely correct model for hypoxia, as it aberrantly induces various hydroxylases not involved in hypoxia pathways and fails to induce downstream biochemical pathways normally induced by hypoxia.
Caki-1 human clear cell renal carcinoma cells were obtained from American Type Culture Collection (ATCC). Caki-1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (GIBCO). For hypoxia treatment, we placed cells into a CO2 incubator with O2 control (BINDER CO2 CB 53) with a regulated environment of 1% O2, 5% CO2 and 94% N2, or cobalt chloride (CoCl2, Sigma) 300 mkM (stock solution 100mM in water) for 24 h.
Total RNA was extracted from Caki-1 cells with Trisol reagent according to the manufacturer’s instructions (Invitrogen). Quality was checked with BioAnalyser and RNA 6000 Nano Kit (Agilent). PolyA RNA was purified with Dynabeads® mRNA Purification Kit (Ambion). An Illumina library was made from polyA RNA with NEBNext® mRNA Library Prep Reagent Set (NEB) according to the manual. Sequencing was performed on HiSeq1500 with 50 bp read length. 10 million reads were generated for each sample.
Reads were mapped to hg19 genome (bowtie2-indexed reference downloaded from ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/hg19.zip) with tophat2 software (version 2.1.0) 14 . Gene models of non-overlapping exonic fragments (http://www-huber.embl.de/pub/DEXSeq/analysis/encode/hsa.DEXSeq.gtf) were taken from ENSEMBL 54 database (http://www.ensembl.org/). For each exonic fragment, total coverage by mapped reads in each sample was calculated with bedtools multicov tool (version 2.17.0). Total gene coverage was calculated as a sum of coverages of all non-overlapping exonic fragments of a gene. Differential expression analysis was performed by applying default read count normalization (estimateSizeFactors) and performing per-gene negative binomial tests (nbinomTest), implemented in DESeq R package (version 1.22.0), with default parameters 15 .
We considered a gene to be differentially expressed if the adjusted p-value in DESeq test was lower than 0.05 and fold-change values were higher than 2 (or lower than ). These sets of differentially expressed genes were further used for gene category enrichment analysis. We took the subset of genes which were found differential in both hypoxia against normoxia controls and after CoCl2 treatment against normal control and only in one of each experiments. These 3 sets of genes were analyzed with DAVID web service (version 6.7) 16 to find KEGG 17 pathways enriched with the genes.
TCGA data on transcriptomes of kidney tumours (KIRC cohort) was downloaded from Broad Institute FireBrowse (http://gdac.broadinstitute.org/runs/stddata__2015_11_01/data/KIRC/20151101/gdac.broadinstitute.org_KIRC.Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.Level_3.2015110100.0.0.tar.gz). Principal component analysis (PCA) was performed with R prcomp function.
A simple transcriptome-based hypoxia signature was constructed as follows: for every sample being evaluated (e.g., TCGA cancer sample), we considered only the genes which were differentially expressed between hypoxia and untreated Caki-1 cell line (DESeq test adjusted p-value< 0.05). For these genes, we multiplied their logarithmic fold-change (hypoxia vs untreated) to their expression in the evaluated sample. The resulting values were then summed up over the genes under consideration. This yielded a per-sample hypoxia score which would be higher in samples with increased expression of hypoxia-induced genes and decreased expression of hypoxia-suppressed genes.
To compare transcriptional effects caused by hypoxia and by CoCl2 exposure, we performed transcriptome sequencing (RNA-seq) of Caki-1 clear cell renal carcinoma cell line in three conditions: untreated, treated with CoCl2, and exposed to hypoxic conditions (1% O2). We searched for differentially expressed genes in two comparisons: CoCl2-treated against untreated cells and cells under hypoxia against untreated cells.
We first asked how transcriptomic changes after both treatments fit into the established hypoxia gene signature 1,18,19 . Figure 1A shows relative expression of the genes from hypoxia signature in all sequenced samples. We observed that genes in hypoxia signature were upregulated both in hypoxia and after CoCl2 treatment, though the effect was much stronger in CoCl2-treated samples. To check if a higher magnitude of transcriptomic changes in CoCl2 compared to hypoxic conditions was characteristic to other differentially expressed genes, we compared the distributions of logarithmic fold-changes (absolute value) for gene expression in these two experiments (Figure 1B). Surprisingly, for the majority of genes, hypoxia-induced changes were higher than the ones induced by CoCl2. In other words, hypoxia resulted in broader transcriptome response than CoCl2 treatment even though specific hypoxia-related genes were more affected after CoCl2 treatment.
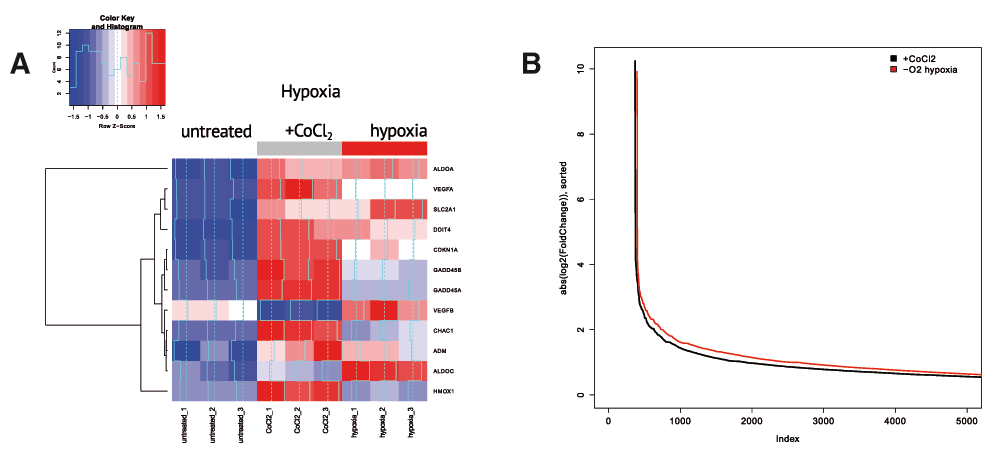
(A) CoCl2 causes much more pronounced expression changes in the expression of key hypoxia regulators compared to real hypoxia treatment. Heatmap represents relative gene expression for key genes involved in hypoxia regulation. (B) Hypoxia results in broader transcriptome response compared to CoCl2 treatment, i.e., more genes are changing expression under hypoxia. The figure shows absolute log fold change values for gene expression between hypoxia (or CoCl2) groups relative to control group. Genes are sorted according to absolute log fold change values. P(wilcoxon) < 2.2 × 10-16.
These results suggested that CoCl2 treatment could be an incomplete model of hypoxia capturing only upstream signalling events in hypoxia pathways and not reflecting broader downstream effects. To further investigate the differences between CoCl2 model and real hypoxia, we compared the sets of differentially up- and down-regulated genes in CoCl2-treatment (against untreated) and in hypoxia-treated (against untreated) cells. Figure 2A summarizes the overlap between those gene sets. To understand what regulatory and biochemical pathways were affected in each treatment, we performed gene category enrichment analysis over KEGG 17 pathways with DAVID web service (version 6.7) 16 for genes affected in both treatments or exclusively in one treatment.
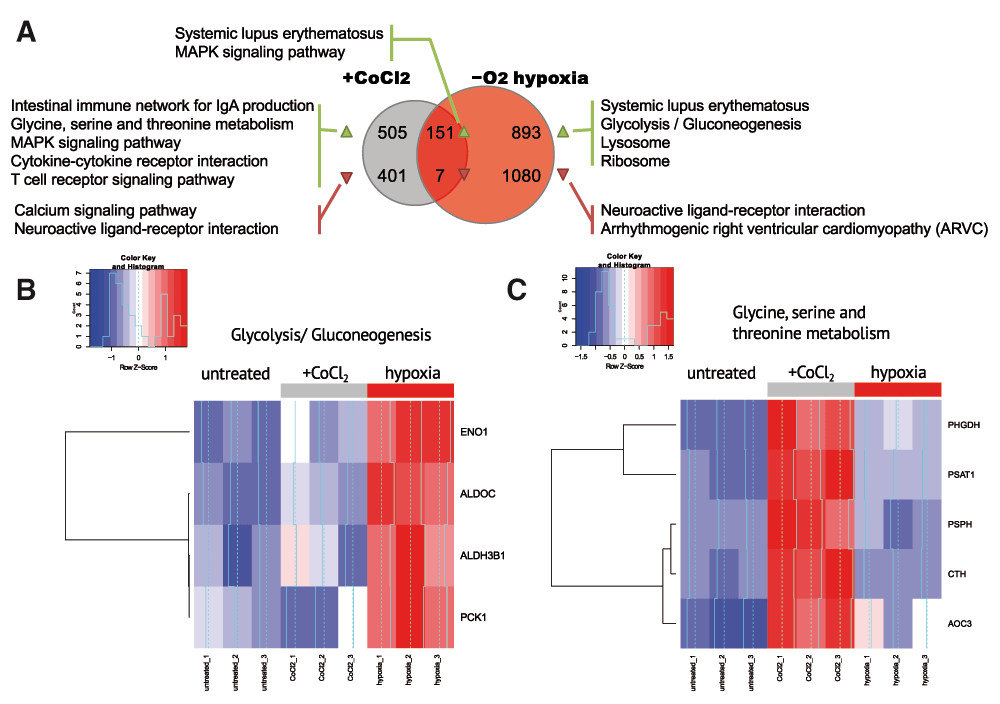
(A) Summary of KEGG pathways, enriched by the genes, up- and down-regulated in CoCl2 and hypoxia treatment. No enriched pathways were discovered for the genes downregulated in both treatments. (B) Overall expression change in glycolysis/gluconeogenesis (KEGG hsa00260) genes in control, hypoxia conditions and after CoCl2 treatment. Glycolysis/Gluconeogenesis (KEGG hsa00010) is activated in hypoxia but not after CoCl2 treatment. (C) Expression change for glycine, serine and threonine metabolism genes (KEGG hsa00010). Glycine, serine and threonine metabolism (KEGG hsa00260) is activated only after CoCl2 treatment but not under hypoxia.
The genes which were significantly upregulated in hypoxic conditions (1% O2) but not after treatment with CoCl2 were significantly enriched in the glycolysis/gluconeogenesis pathway which was known to be related to hypoxia 1,15,18 . Unexpectedly, we detected no enrichment in glycolysis/gluconeogenesis pathway with the genes differentially expressed after CoCl2 treatment which was confirmed by a heatmap for glycolysis/gluconeogenesis-related genes (Figure 2B).
The genes upregulated in both hypoxia and under CoCl2 treatment were enriched in the MAPK pathway. As the MAPK pathway was known to activate hypoxic response, MAPK activation was expected in hypoxia. Even though CoCl2 affected directly HIF1a pathway, MAPK was activated in CoCl2-treated samples as well as in real hypoxia. This result supported a previous observation of MAPK-dependent activation of hypoxia response under CoCl2 treatment 12 . Surprisingly, we observed systemic lupus erythematosus-related pathway activation in both treatments. The set of genes upregulated in this pathway (histone proteins H2A, H2B, H3, H4 and MHCII antigen-presenting genes) could be unrelated to lupus erythematosus, but rather could indicate increased proliferation and inflammation.
We also explored the genes specific to CoCl2 treatment but not affected by hypoxia. Surprisingly, the genes upregulated after CoCl2 treatment but not changed in hypoxia were enriched in the glycine/serine/threonine biosynthesis pathway (Figure 2A and 2B). We hypothesized that Co2+ ion could substitute metal cofactors of several oxidoreductases in the pathway and subsequently impair their activity. This, in turn, could require greater amounts of enzyme to be synthesized.
Hypoxic conditions within a tumour have been shown to predict worse clinical outcome 20 . We used our data on whole-transcriptome profiling of kidney cancer cell lines in normal and hypoxic conditions to extract a wide hypoxia signature and validate it with TCGA data on transcriptomes of kidney tumours 21 . Major variation in TCGA transcriptomes (Figure 3A) is generated by the difference between tumours and adjacent normal samples. We projected our sequenced samples to the principal components derived from TCGA samples and observed that the direction of transcriptome changes between untreated and hypoxic cell lines is slightly similar to the difference between normal and tumour samples, though hypoxia-related changes couldn’t explain normal-tumour difference. We constructed a transcriptome-based hypoxia signature as described in the Methods section. To test if this hypoxia signature predicted clinical outcome for kidney cancer patients, we explored the distribution of our hypoxia scores between disease free patients and patients in which the disease had recurred or progressed. Hypoxia scores were significantly higher for recurred/progressed patients (Wilcoxon test p-value=0.0009, Figure 3B).
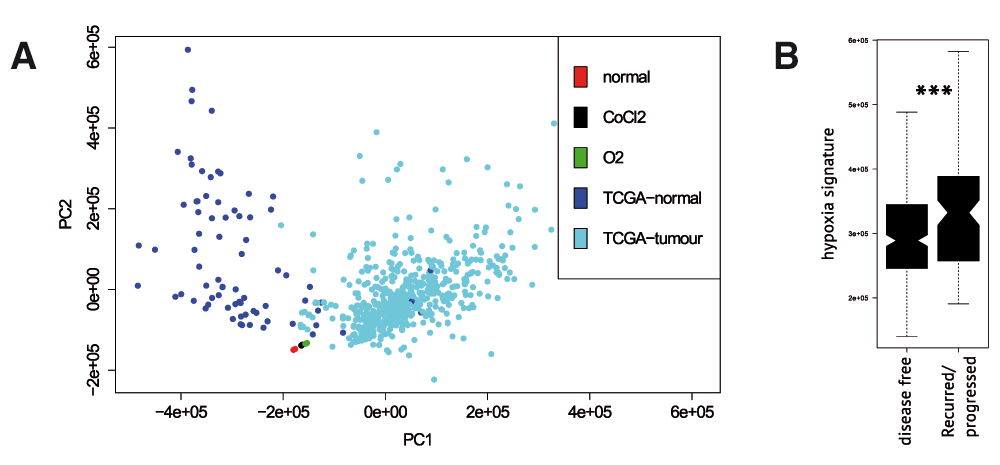
(A) PCA plot for TCGA samples of kidney tumours, adjacent normal tissue samples, untreated Caki-1 cells, Caki-1 cells treated with CoCl2 and the cells in hypoxic environment. (B) Hypoxia signature derived from RNA-seq results predicted significantly higher hypoxia scores for recurred or progressed TCGA tumours.
The current study for the first time provides RNA-seq data revealing hypoxia-induced transcriptomic changes, which allows broader understanding of the processes related to hypoxia. In our analysis, we explored the limits of applicability of CoCl2 treatment as the model of hypoxia. Briefly, we observed that CoCl2 strongly alters expression of few genes important for hypoxia signalling, but fails to influence the essential downstream consequences of hypoxia, particularly the glycolysis/gluconeogenesis pathway. This might suggest the existence of alternative regulation mechanisms which trigger the downstream events in hypoxia together with main VHL/HIF1a pathway. CoCl2 treatment also abberantly induced pathways which did not respond to hypoxia. These included the glycine, serine and threonine metabolism pathways. We hypothesized that its aberrant activation might be caused by Co2+ ion binding to the enzymes (other than PHD proteins) involved in Glycine, Serine and Threonine biosynthesis.
F1000Research: Dataset 1. Raw per-gene expression counts for individual genes (see Methods), 10.5256/f1000research.7571.d109570 23
RNA-seq data was deposited to NCBI SRA under SRP066934 study accession code. The study contained experiments under the following accession codes: untreated (SRX1459966, SRX1459967, SRX1459969), treated with CoCl2 (SRX1459974, SRX1459977, SRX1459978), exposed to hypoxia (SRX1459979, SRX1459981, SRX1459984).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)