Keywords
Oxidative damage, mitochondrial DNA repair, mitochondrial pathology, mitochondrial mutations
Oxidative damage, mitochondrial DNA repair, mitochondrial pathology, mitochondrial mutations
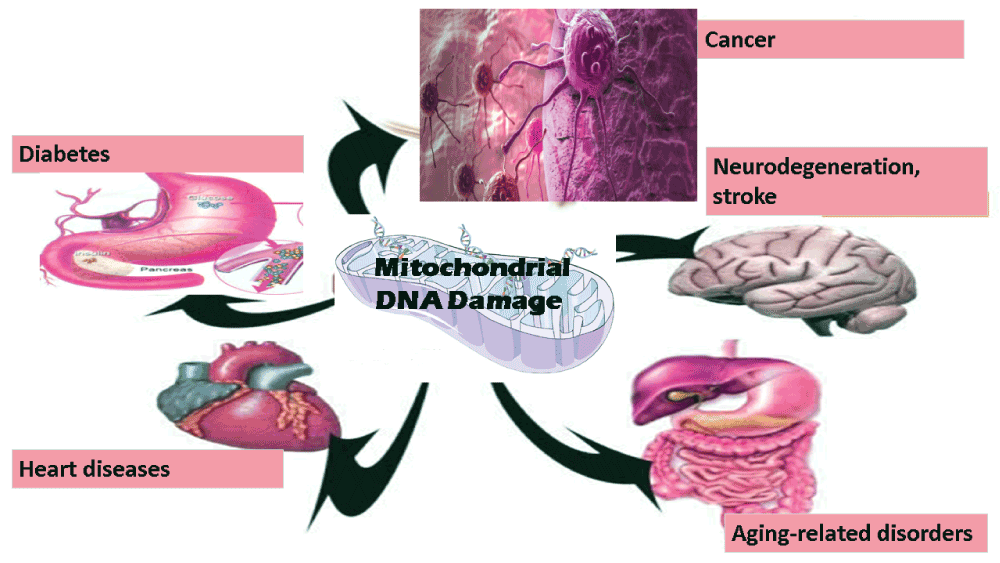
Mitochondria, a key organelle of most eukaryotic cells, are not only essential for cellular energy generation but also important for calcium metabolism and apoptotic cell-signaling1. Like the nucleus, both mitochondria and chloroplasts contain their own DNA, and mitochondrial DNA (mtDNA) damage has been frequently implicated in several diseases including neurodegeneration, cancer, stroke, cardiomyopathy, diabetes, and aging-related disorders (2, Figure 1). Unlike nuclear DNA, the mitochondrial genome is circular, contains very few introns, and the number of mtDNA copies in one mitochondrion can be in the range of two to ten. Furthermore, the size of mtDNA is very small (16.6 kb in humans), and mitochondrial codon-usage is also different. The multicopy nature of mtDNA bestows unconventional modes of DNA maintenance such as selective degradation of damaged DNA, and an unusual form of recombination3. mtDNA is maternally inherited, and sperm mitochondria are mostly degraded after fertilization4. Mitochondria synthesize some of its own proteins, and one of the reasons for this could be that all proteins that are translated in cytoplasm might not be able to cross mitochondrial membranes owing to their varied hydrophobicity5. mtDNA encodes 22 tRNAs, 2 rRNAs, and 13 proteins that participate in mitochondrial ATP synthesis6. Reactive oxygen species (ROS) are very reactive oxygen-containing molecules. ROS are produced in all types of cells and can have various harmful effects. mtDNA, like other DNA, can not only be damaged by radiation and genotoxic chemicals but also by ROS that are frequently produced in mitochondria7. mtDNA damage can exaggerate further because of errors during DNA replication, and lack of conventional histone proteins in mitochondria8. ROS can cause various types of oxidative damage including DNA strand breaks, base modification or removal, and cross linking. DNA polymerase γ (pol γ), the only DNA polymerase known to be present in the mitochondria, have low frameshift fidelity, and, is believed to be a major contributor to changes in mtDNA9.
Several studies report the effect of genotoxic agents on mitochondria10,11. However, it is not easy to draw conclusions in these cases, as agents that damage mtDNA also damage nuclear DNA. Therefore, it is suggested that all studies should compare consequences of nuclear and mtDNA damage in such cases, as far as possible. Other than its involvement in cancer and neurological disorders, changes in mtDNA have been shown to be associated with a few hereditary diseases12. mtDNA damage is well known to cause impaired bioenergetics, reduced cell proliferation and apoptosis, hypercholesterolemia, and atherosclerosis12. Interestingly, mtDNA defects are known to cause defective mitochondrial ATP generation that results into compromised organ function and diseases13.
In case of the most common neurodegenerative disorders including Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS) also, mtDNA damage has been implicated as a factor that cause or exaggerate these diseases14. Brain tissues from Alzheimer's patients show greater fragmentation mtDNA. However, similar damage to nuclear DNA is controversial in this case. Increased mtDNA damage was also associated with reduced levels of mitochondrial protein expression13. Interestingly, brain tissues from Alzheimer's patients show higher levels of oxidized bases. In this case, mtDNA was found to have 10-times more oxidized bases compared to nuclear DNA indicating that mtDNA is more succeptible to oxidants14.
In the case of Huntington disease (HD), higher levels of oxidative stress were observed in the brain tissues of both humans and mice16. In the case of a mouse model of HD, embryonic fibroblasts showed increased mitochondrial matrix Ca2+ loading, and higher superoxide generation. This confirmed that both mitochondrial Ca2+ signaling and superoxide generation are dysregulated in HD, and, reducing mitochondrial Ca2+ uptake can be a therapeutic strategy for HD16. Peripheral blood mononuclear cells (PBMCs) from systemic lupus erythematosus (SLE) patients also exhibited enhanced mtDNA damage indicating potential role of mitochondria in the pathogenesis of SLE17. Apolipoprotein E (ApoE) is known to play a protective role in preventing artery wall thickening in atherosclerosis and ApoE-/- mice show mtDNA damage before significant atherosclerosis18. pol γ-/-/ApoE-/- mice show extensive mtDNA damage, impaired mitochondrial respiration, and increased atherosclerosis, even without increased ROS. Furthermore, pol γ-/-/ApoE-/- monocytes showed increased inflammatory cytokine release18. Aging is often associated with the accumulation of deleterious changes, reduced physiological functions, and increased likelihood of diseases19. In this context, a number of mitochondrial aberrations have been observed with aging. These aberrations are accumulation of mtDNA mutation, inefficient oxidative phosphorylation, increased production of ROS, and disorganized mitochondrial structure20. These mtDNA mutations are often somatic, with variable changes in individual cells. Often, higher levels of these mutations are associated with respiratory chain deficiency. A mosaic pattern of respiratory chain deficiency can be found in different tissues because of uneven distribution of mutations13. The mitochondrial free radical theory of aging has been one of the most supported ideas of aging19. This theory postulates that the production of intracellular ROS is the major determinant during aging. Several invertebrate and mammalian models already support this hypothesis. Oxidative stress, when propagated by active radicals, can damage DNA, phospholipids, proteins and other biomolecules. Reactive oxygen species mediated mtDNA damage can occur directly at the sugar-phosphate backbone, at the bases, or in the form of single and double strand breaks20. Unfortunately, most of the antioxidant-supplementation regimens do not increase longevity, as predicted by the free radical theory of aging. Intracellular ROS are generated in multiple compartments and by multiple pathways. Important contributors in this case are NADPH oxidases, cyclooxygenases, and lipid metabolism enzymes21. Despite several non-mitochondrial contributors, almost 90% of cellular ROS are still generated in mitochondria. In some cases, long-lived species were not only found to produce less ROS but also showed less oxidative damage22. Similarly, various animal and human studies suggest that the decline in muscle mitochondria is a leading factor for muscular abnormalities23.
Aged monkeys showed enhanced DNA damage and reduced transcription of mtDNA compared to young ones24. D-gal-induced aging rats are important animal model of aging, and the level of mtDNA deletions was found to be significantly more in the hippocampus of D-gal-treated rats compared to controls25. NADPH oxidase (NOX) generates ROS while transporting electrons across the mitochondrial membrane. Similarly, uncoupling protein 2 (UCP2) transports anions and protons across the mitochondrial membrane, and also controls ROS generation. In case of D-gal-induced animal model of aging, damaged mitochondrial ultrastructure was seen in the hippocampus region along with increased production of NOX and UCP2. Nicotinamide adenine dinucleotide (NAD+) is a key electron transporter in mitochondria. NAD+ depletion may play a prominent role in the aging process, not only by limiting energy production, but also by compromising DNA repair and genomic signaling as NAD+ is an important substrate for the nuclear repair enzymes21. Poly(ADP-ribose) polymerase (PARP) controls inflammatory immune responses, and hyperactivation of PARP-1 is known to activate mitochondrial pathway of apoptosis26. Age-associated increase in oxidative nuclear damage was found to be associated with PARP-induced NAD+ depletion and absence of SIRT1 activity in rodents26. Ercc1 mutant mice, which are deficient in DNA repair pathways, show accelerated aging and progressive memory loss27. Defective oxidative phosphorylation, mutated mtDNA, or mitochondrial ROS have also been documented in cases of tumorigenesis28. Oxidative stress in the cardiovascular system is known to cause accumulation of reactive oxygen and nitrogen species, which increase leukocyte adhesion and endothelial permeability29. NFκB is one of the most important transcription factor that is known to be involved in important signaling pathways, development, and several diseases. Hypoxia-Inducible Factor (HIF-1) is a protein that not only protects from hypoxia-induced damage, but is also important for smooth functioning of immune system and key metabolic pathways. In an interesting study, ROS, NFκB- and HIF1-activation in the tumor microenvironment induced accelerated aging in rodents, which subsequently caused stromal inflammation and altered cancer cell metabolism30. Certain dietary treatments or enrichment of mitochondrial membranes with oxidant-resistant fatty acids were found to increase life span in rodents31. Monounsaturated-fatty-acid-rich diet prevented the accelerated mtDNA mutations in the brain mitochondria from aged animals. Therefore, changes in mtDNA that gradually accumulate in a variety of tissues during aging appear to be involved in onset of various diseases32 and a better understanding of mitochondrial biology is required in this perspective. mtDNA ligase is essential for cell survival particularly because of its role in base excision repair pathway33.
Mitochondria are of central importance in eukaryotic cells. However, mtDNA is more prone to damage, and mtDNA repair pathways are inadequate. Together, these problems might frequently lead to unrepaired mtDNA lesions, and defective energy metabolism. mtDNA damage has been frequently shown to be involved in initiation and progression of several diseases including various types of neurodegenerative disorders, cancer, stroke, heart-diseases, and diabetes. There is an urgent need for detailed investigation in this area, to find out the mitochondrial contribution to various diseases, so that improved prevention measures and cures can be developed.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)